Medisch expert van het artikel

Nieuwe publicaties
Angelman syndroom bij kinderen en volwassenen
Laatst beoordeeld: 04.07.2025

Alle iLive-inhoud wordt medisch beoordeeld of gecontroleerd op feiten om zo veel mogelijk feitelijke nauwkeurigheid te waarborgen.
We hebben strikte richtlijnen voor sourcing en koppelen alleen aan gerenommeerde mediasites, academische onderzoeksinstellingen en, waar mogelijk, medisch getoetste onderzoeken. Merk op dat de nummers tussen haakjes ([1], [2], etc.) klikbare links naar deze studies zijn.
Als u van mening bent dat onze inhoud onjuist, verouderd of anderszins twijfelachtig is, selecteert u deze en drukt u op Ctrl + Enter.

Er zijn een aantal ziekten waarvoor uitdrukkingen als "zorg goed voor jezelf en je wordt niet ziek" op zijn minst belachelijk klinken. Dit zijn pathologieën waarbij bepaalde psychische en lichamelijke afwijkingen al vóór de geboorte in het lichaam van het kind aanwezig zijn, maar de ouders hiervoor niet verantwoordelijk zijn. Dergelijke ziekten worden veroorzaakt door mutaties of afwijkingen in chromosomensets en worden chromosomaal of genetisch genoemd. Het Angelman-syndroom, het Down-syndroom, het Patau-syndroom, het Edwards-syndroom, het Turner-syndroom, het Prader-Willi-syndroom - dit is slechts een deel van de genetische ziekten uit een behoorlijk lange lijst.
Happy Man Syndroom
Deze keer hebben we het over de pathologie die vernoemd is naar de Engelse kinderarts Harry Angelman, die dit probleem voor het eerst in 1965 aankaartte. Hij had de dag ervoor drie ongewone kinderen in zijn praktijk gezien, die allemaal dezelfde, eigenaardige symptomen vertoonden. De arts noemde deze kinderen poppenkinderen en schreef er een artikel over, dat aanvankelijk "Kinderen-marionettes" heette. Het artikel zelf en de titel ervan waren geschreven in de veronderstelling dat er een schilderij te zien was in een van de musea van Verona. Het schilderij toonde een lachende jongen en heette "De Poppenjongen". De associatie van het kind op het schilderij met de drie kinderen die Angelman ooit in zijn praktijk had gezien, bracht de kinderarts ertoe de kinderen vanwege de ziekte in één groep samen te voegen.
Het is niet verwonderlijk dat de in het artikel genoemde kinderen niet door andere artsen zijn opgemerkt. Op het eerste gezicht leken ze immers totaal verschillende ziekten te hebben, zo verschillend was het algemene klinische beeld van de ziekte in drie verschillende gevallen. Misschien zou de "nieuwe" chromosomale pathologie andere wetenschappers hebben geïnteresseerd, maar de genetica was destijds nog niet ver genoeg ontwikkeld om de hypothese van de Engelse arts te bevestigen. Daarom belandde het artikel, na enige interesse erin, lange tijd op de plank.
De volgende vermelding van het Angelman-syndroom, zoals het artikel van de Engelse kinderarts G. Angelman nu heette, dateert uit het begin van de jaren 80 van de 20e eeuw. Pas in 1987 kon de reden worden gevonden waarom een klein deel van de kinderen met zulke afwijkingen wordt geboren dat ze van buitenaf constant glimlachend en blij lijken. In werkelijkheid is dit helemaal niet waar; de glimlach is slechts een grimas, waarachter een ongelukkige menselijke ziel en de pijn van de ouders schuilgaan.
Epidemiologie
Volgens de statistieken kan een chromosomale mutatie bij een kind zich zowel ontwikkelen tegen de achtergrond van vergelijkbare mutaties bij de ouders als zonder dergelijke mutaties. Er is geen duidelijke erfelijke aard van het Angelman-syndroom (AS), maar de kans op het ontwikkelen van pathologie bij ouders met chromosomale mutaties is vrij groot.
Interessant is ook dat als er in een gezin al een kind met AS is, de kans één procent is dat een tweede kind dezelfde stoornis krijgt, zelfs als de ouders gezond zijn.
Er zijn nog steeds geen exacte statistieken over het aantal patiënten met het Angelman-syndroom. Mogelijk is de reden hiervoor de verscheidenheid aan symptomen, die in een bepaalde samenstelling kunnen voorkomen of lange tijd helemaal niet kunnen optreden. Aangenomen wordt dat de prevalentie van de ziekte 1 kind per 20.000 pasgeborenen is. Maar dit cijfer is zeer bij benadering.
Oorzaken Angelman syndroom
Het Angelmansyndroom is een medische naam voor een chromosomale pathologie, maar het is lang niet de enige. Mensen noemen deze ziekte het poppenkindersyndroom, het blijpoppensyndroom, het Petrushkasyndroom en het lachpoppensyndroom. Mensen verzinnen allerlei namen (soms zelfs beledigend voor de patiënten zelf en hun ouders), maar een ziekte is een ziekte, hoe grappig die er ook uitziet en wat de redenen ook zijn.
En de oorzaken voor de ontwikkeling van het Angelman-syndroom, net als bij veel andere genetische aandoeningen, zijn in alle gevallen verstoringen in de structuur van een van de chromosomen of de chromosomenset als geheel. Maar in ons geval ligt het hele probleem bij chromosoom 15, dat van de moeder wordt doorgegeven. Dat wil zeggen dat het vaderlijke chromosoom in dit geval geen afwijkingen vertoont, maar dat het vrouwelijke chromosoom bepaalde mutaties ondergaat.
Afhankelijk van het type chromosomale afwijking wordt het Angelman-syndroom geclassificeerd als een chromosomale mutatie. Dergelijke mutaties worden beschouwd als:
- Een deletie (het ontbreken van een deel van een chromosoom dat een bepaalde set genen bevat; als een van de genen ontbreekt, spreken we van een microdeletie) is het gevolg van twee breuken en één hereniging, waarbij een deel van het oorspronkelijke chromosoom verloren gaat.
- Duplicatie (de aanwezigheid van een extra deel in een chromosoom dat een kopie is van een bestaand deel) leidt in de meeste gevallen tot de dood van een persoon en minder vaak tot onvruchtbaarheid.
- Inversie (omkering van één van de delen van het chromosoom met 180 graden, d.w.z. in de tegenovergestelde richting, en dan liggen de genen daarop in de tegenovergestelde volgorde). Dit houdt in dat de gebroken uiteinden van het chromosoom in een andere volgorde dan in de oorspronkelijke volgorde aan elkaar worden verbonden.
- Insertie (als een deel van het genetische materiaal in een chromosoom niet op zijn plaats zit),
- translocatie (als een bepaald deel van een chromosoom aan een ander chromosoom vastzit; zo'n mutatie kan wederzijds zijn zonder verlies van delen).
Wanneer een kind een gemuteerd chromosoom ontvangt van een nietsvermoedende moeder, is het gedoemd om met afwijkingen geboren te worden. De meest voorkomende oorzaak van het Angelman-syndroom wordt nog steeds beschouwd als een deletie van het 15e chromosoom van de moeder, waarbij een klein stukje ontbreekt. Minder vaak voorkomende mutaties bij het "lachpop"-syndroom zijn:
- translocatie,
- unipaternale disomie (als het kind een paar chromosomen van de vader heeft ontvangen, ontbreekt het moederlijke chromosoom),
- mutatie van genen in het DNA, die zowel het belangrijkste bouwmateriaal (genetisch materiaal) als de instructies voor het juiste gebruik ervan vormen (in het bijzonder mutatie van het ube3a-gen op het materneel chromosoom).
De aanwezigheid van een van deze mutaties bij de ouders is een risicofactor voor de ontwikkeling van het Angelman-syndroom bij kinderen. Maar niet alleen chromosomale mutaties, maar ook genomische mutaties (die gepaard gaan met een kwantitatieve verandering in chromosomensets en vaker voorkomen dan chromosomale mutaties) kunnen de ontwikkeling van de ziekte bij een kind veroorzaken. Een veelvoorkomende genomische mutatie is chromosoomtrisomie (wanneer iemands chromosomenset meer dan 46 chromosomen bevat).
Om een pathologie bij een kind te laten zien, is het helemaal niet nodig dat de ouders chromosomale afwijkingen hebben. Toch is er een bepaald percentage patiënten bij wie de ziekte erfelijk is.
Pathogenese
Laten we wat dieper ingaan op de biologie, of preciezer gezegd, genetica. De genetische informatie van elk individueel menselijk organisme is vervat in 23 chromosomenparen. Eén chromosoom van een paar wordt door de vader aan het kind doorgegeven, het andere door de moeder. Alle chromosomenparen verschillen in vorm en grootte en dragen bepaalde informatie met zich mee. Zo is het 23e chromosomenpaar (X- en Y-chromosomen) verantwoordelijk voor de vorming van de geslachtskenmerken van de baby (XX - meisje, XY - jongen, terwijl het Y-chromosoom het kind alleen van de vader kan ontvangen).
Idealiter krijgt een kind 46 chromosomen van zijn ouders, die zijn genetische kenmerken vormen en hem als individu bepalen. Een groter aantal chromosomen wordt trisomie genoemd en wordt beschouwd als een afwijking van de norm. Zo kan de aanwezigheid van chromosoom 47 in de chromosomenset (karyotype, bepalend voor soort en individuele kenmerken) het syndroom van Down veroorzaken.
Als de chromosomen met een speciale kleurstof worden gekleurd, kun je onder de microscoop strepen van verschillende tinten langs elk ervan zien. Binnen elke streep bevindt zich een enorm aantal genen. Al deze strepen zijn door wetenschappers genummerd en hebben een vaste locatie. De afwezigheid van een van de strepen wordt beschouwd als een afwijking van de norm. Bij het Angelman-syndroom is de afwezigheid van segmenten van het moederlijke chromosoom in het interval q11-q13, gelegen in de lange arm, waar het aantal DNA-basen slechts ongeveer 4 miljoen bedraagt, zeer vaak te zien.
Het hoofdbestanddeel van het chromosoom wordt beschouwd als een ongelooflijk lang DNA-molecuul met duizenden genen en tientallen en honderden miljoenen stikstofbasen. Zo bevat chromosoom 15, verantwoordelijk voor de ontwikkeling van het Angelman-syndroom en diverse andere aandoeningen, 1200 genen en ongeveer 100 miljoen basen. Elke verstoring in de structuur van het DNA-molecuul zal zeker van invloed zijn op het uiterlijk en de ontwikkeling van het toekomstige kind.
De genetische informatie in genen wordt omgezet in eiwit of RNA. Dit proces wordt genexpressie genoemd. Zo krijgt de genetische informatie die ouders van hun ouders ontvangen zowel vorm als inhoud, die wordt belichaamd in hun unieke vrouwelijke of mannelijke erfgenaam.
Er bestaan een aantal pathologieën met een niet-klassieke vorm van overerving, waaronder het Angelman-syndroom. Hierbij worden genen die via gepaarde chromosomen van de ouders worden doorgegeven, op een unieke manier geïmpliceerd door de ouders en zich op verschillende manieren manifesteren.
Het Angelman-syndroom is dus een treffend voorbeeld van genomische imprinting, waarbij de genexpressie in het lichaam van het kind direct afhankelijk is van de ouder van wie de allelen zijn geërfd (verschillende vormen van één gen, geërfd van vader en moeder, gelegen op identieke delen van gepaarde chromosomen). Dat wil zeggen dat alleen afwijkingen op het moederlijke chromosoom leiden tot de ontwikkeling van het syndroom, terwijl mutaties en structurele stoornissen op het vaderlijke chromosoom volledig andere pathologieën veroorzaken.
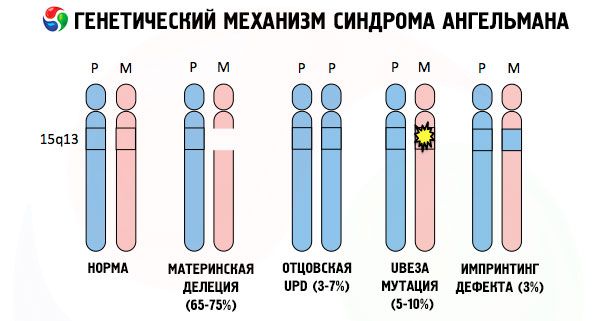
Bij deze pathologie is er sprake van een tekort aan bepaalde genen op het moederlijk chromosoom of een verlies/vermindering van de activiteit van individuele genen (in de overgrote meerderheid van de gevallen het ube3a-gen, dat betrokken is bij de stofwisseling van ubiquitine, een eiwit dat de afbraak van andere eiwitten reguleert). Als gevolg hiervan worden bij het kind geestelijke ontwikkelingsstoornissen en lichamelijke misvormingen vastgesteld.
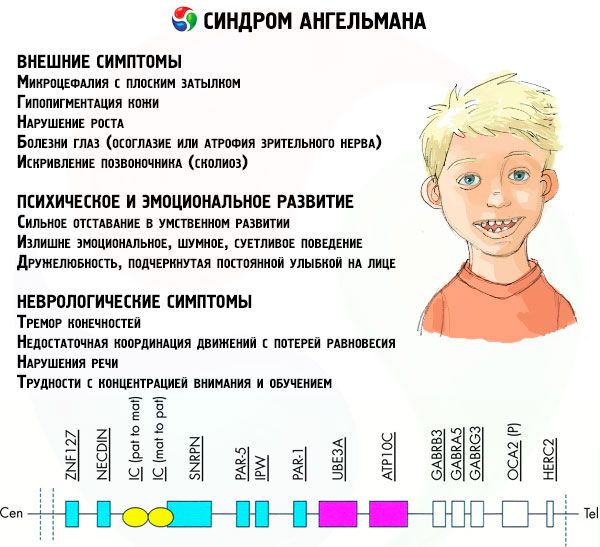
Symptomen Angelman syndroom
De symptomen van het Angelman-syndroom beïnvloeden verschillende aspecten van het leven en de ontwikkeling van een kind: fysiek, neurologisch en mentaal. Op basis hiervan kunnen drie groepen symptomen worden onderscheiden die wijzen op de ontwikkeling van deze pathologie.
- Uitwendige of lichamelijke symptomen:
- een onevenredig klein hoofd in vergelijking met het lichaam en de ledematen, die van normale grootte zijn,
- te brede mond,
- er is bijna altijd een glimlach op het gezicht (met een open mond),
- schaarse tanden,
- smalle bovenlip,
- vaak uitstekende brede tong,
- uitstekende onderkaak,
- puntige kin,
- zeer lichte huid, vaak behaard (albinisme, geassocieerd met het feit dat het lichaam het pigment melanine niet aanmaakt),
- donkere vlekken op een lichte huid (hypopigmentatie als gevolg van onvoldoende melanineproductie)
- fysieke of externe symptomen: oogziekten zoals scheelzien of atrofie van de oogzenuw,
- kromming van de wervelkolom (scoliose),
- stijve benen (bij het lopen buigt een persoon zijn benen niet bij de knieën vanwege de beperkte mobiliteit van de gewrichten, vandaar de vergelijking met de gang van een pop).
- Symptomen gerelateerd aan mentale en emotionele ontwikkeling:
- ernstige mentale retardatie,
- overdreven emotioneel, luidruchtig, kieskeurig gedrag,
- vaak in de handen klappen,
- uitgedrukte vriendelijkheid, benadrukt door een constante glimlach op het gezicht,
- vaak zonder reden lachen.
- Neurologische symptomen:
- tremor van de ledematen,
- onvoldoende coördinatie van bewegingen met verlies van evenwicht,
- verminderde spierspanning,
- verschillende slaapstoornissen,
- frequente hysterische aanvallen in de kindertijd,
- spraakstoornissen (het kind begint laat te praten, heeft slechte communicatievaardigheden en spreekt onduidelijk),
- hyperactiviteit tegen de achtergrond van verhoogde prikkelbaarheid,
- concentratie- en leerproblemen.
Maar dit is een algemeen beeld van de ziekte. Het klinische beeld van het Angelman-syndroom hangt in feite grotendeels af van het ontwikkelingsstadium van de ziekte en het type chromosomale mutatie dat de pathologie heeft veroorzaakt. Dit betekent dat de symptomen van de ziekte aanzienlijk kunnen verschillen bij verschillende patiënten, waardoor het ons lange tijd niet mogelijk was om de pathologie te onderscheiden van anderen met een vergelijkbaar klinisch beeld.
Uit het totale aantal symptomen kunnen we de symptomen benadrukken die voor alle patiënten zonder uitzondering kenmerkend zijn:
- ernstige mentale retardatie,
- ongepast gedrag (onredelijk lachen, verhoogde opwinding, slechte concentratie, staat van euforie),
- onderontwikkeling van motorische vaardigheden,
- slechte coördinatie van bewegingen, loopataxie (onregelmatig tempo, heen en weer zwaaien, enz.), tremor van de ledematen.
- spraakontwikkelingsstoornis waarbij de non-verbale communicatiemiddelen overheersen.
Onder de symptomen die de overgrote meerderheid van de patiënten ervaart, kunnen de volgende worden onderscheiden:
- onevenredigheid tussen het hoofd en het lichaam veroorzaakt door een vertraagde fysieke ontwikkeling,
- bij veel patiënten is de vorm van de schedel zodanig dat de omvang van de hersenen kleiner blijft dan bij gezonde mensen (microcefalie),
- epileptische aanvallen vóór de leeftijd van 3 jaar met een progressieve afname in sterkte en frequentie op oudere leeftijd,
- vervorming van EEG-parameters (schommelingen en hoge amplitude van laagfrequente golven).
Deze symptomen komen vrij vaak voor, maar bij 20% van de patiënten met het Angelman-syndroom zijn ze niet aanwezig.
Nog zeldzamer kunnen de volgende verschijnselen van de ziekte worden vastgesteld:
- ernstig of mild scheelzien,
- slechte controle over de tongbeweging, waardoor patiënten vaak zonder reden hun tong uitsteken,
- moeilijkheden met slikken en zuigen, vooral bij jonge kinderen,
- verstoring van de huid- en oogpigmentatie,
- armen omhoog of gebogen tijdens het lopen,
- hyperreflexie,
- slaapstoornissen, vooral in de kindertijd,
- frequente speekselvloed,
- onverzadigbare dorst,
- overactieve kauwbewegingen,
- overgevoeligheid voor hitte,
- platte achterkant van het hoofd,
- uitstekende onderkaak,
- zachte handpalmen.
Een aanzienlijk deel van de patiënten heeft last van urineproblemen, die ze slecht beheersen, een verminderde fijne motoriek, wat leidt tot problemen met zelfzorg en leervermogen, en overgewicht. Bijna alle patiënten komen later in de puberteit dan gezonde leeftijdsgenoten.
Kinderen met het Angelman-syndroom kunnen gesproken taal goed verstaan en begrijpen, maar willen niet deelnemen aan gesprekken, waardoor hun spraak beperkt blijft tot enkele tientallen woorden die in het dagelijks leven nodig zijn. Op volwassen leeftijd zien deze patiënten er echter jonger uit dan leeftijdsgenoten zonder genetische afwijkingen.
Veel symptomen van het Angelman-syndroom zijn veranderlijk, waardoor het klinische beeld van de ziekte aanzienlijk verandert met de leeftijd. Convulsies en epileptische aanvallen nemen af of verdwijnen helemaal, de patiënt raakt minder opgewonden en de slaap verbetert.
Complicaties en gevolgen
Het Angelmansyndroom is een ernstige, momenteel vrijwel ongeneeslijke chromosomale pathologie die patiënten de mogelijkheid ontneemt een normaal leven te leiden. Hoe het leven van een kind met AS eruitziet, hangt grotendeels af van het type chromosomale afwijking.
Duplicatie van een chromosoomsegment is in de meeste gevallen onverenigbaar met het leven. En zelfs als dergelijke patiënten niet op jonge leeftijd sterven en de puberteit bereiken, hebben ze geen kans om kinderen te krijgen.
De deletie of afwezigheid van een deel van de genen die het vaakst voorkomen bij het Angelman-syndroom, vormt een belemmering voor het kind om te leren lopen en praten. Bij deze kinderen is de verstandelijke beperking ernstiger en komen epileptische aanvallen vaker voor en zijn ze veel heftiger dan bij patiënten met andere chromosomale afwijkingen.
Als er sprake is van slechts een mutatie in één gen, kan het kind met de nodige aandacht en aanpak de basisbeginselen van zelfzorg, communicatie en interactie in een groep worden aangeleerd. Toch zal het in ontwikkeling nog steeds achterlopen op zijn leeftijdsgenoten.
Voor kinderen met het Angelman-syndroom, die van nature vriendelijk zijn, is de liefde en aandacht van hun ouders het allerbelangrijkste. Alleen in dit geval zal de opvoeding van het kind vruchten afwerpen, hoe beperkt ook. Patiënten met AS kunnen natuurlijk niet naar een gewone school. Ze hebben speciale klassen nodig waar kinderen eerst leren zich te concentreren en vervolgens geleidelijk de basis van schoolkennis leren.
Diagnostics Angelman syndroom
Het Angelman-syndroom is een aangeboren ontwikkelingsstoornis. Door bepaalde omstandigheden is het echter vaak onmogelijk om de diagnose al in de zuigelingen- en vroege kindertijd te stellen. Dit komt door de aspecificiteit en zwakke expressie van symptomen bij zuigelingen en kinderen jonger dan 3 jaar. Bovendien is de prevalentie van de ziekte in ons land niet zo groot dat artsen deze onder leeftijdsgenoten hebben leren herkennen.
Het Angelman-syndroom bij baby's kan zich uiten in een verminderde spierspanning, wat zich uit in problemen met de voeding (zwakte van de zuig- en slikreflex) en later in moeilijkheden met het leren lopen (dergelijke kinderen beginnen veel later te lopen). Deze symptomen zijn de eerste tekenen van een ontwikkelingsafwijking bij de baby, die mogelijk samenhangt met een chromosoomafwijking. Alleen genetische analyse kan deze aanname bevestigen.
Speciale aandacht wordt besteed aan kinderen van ouders met diverse genomische of chromosomale afwijkingen. De ziekte manifesteert zich immers mogelijk niet meteen, en als de pathologie tijdig wordt ontdekt, kan door intensieve begeleiding met het kind aanzienlijk meer leerprestaties worden behaald, waardoor de progressie van de ziekte wordt vertraagd.
Als de ouders verschillende chromosomale afwijkingen hebben, wordt er nog vóór de geboorte van het kind een genetische analyse uitgevoerd, omdat SA een van de pathologieën is die al in het embryonale stadium kan worden opgespoord.
Het verzamelen van materiaal voor genetisch onderzoek kan op twee manieren plaatsvinden:
- invasief (met een bepaald percentage risico, aangezien het nodig is de baarmoeder te penetreren om een monster vruchtwater te nemen),
- niet-invasief (analyse van het DNA van de baby uit het bloed van de moeder).
Vervolgens worden de volgende onderzoeken uitgevoerd:
- fluorescerende in situ-hybridisatie (FISH-methode) – binding van een DNA-sonde die is gelabeld met een speciale kleurstof aan het te bestuderen DNA, gevolgd door onderzoek onder een microscoop.
- analyse van mutaties in het ube3a-gen en geïmprinte genen,
- DNA-methyleringsanalyse met behulp van speciale methoden die in de genetica worden gebruikt.
Genetische tests geven vrij nauwkeurige informatie over chromosomale afwijkingen, waardoor toekomstige ouders vooraf weten waar ze zich op moeten voorbereiden. Er zijn echter uitzonderingen. Bij een bepaalde groep patiënten, met alle symptomen die wijzen op een pathologie, blijven de testresultaten normaal. Dat wil zeggen dat pathologie alleen kan worden vastgesteld door het kind vanaf de vroege kinderjaren zorgvuldig te observeren: hoe het eet, wanneer het begon te lopen en te praten, of het zijn benen buigt tijdens het lopen, enzovoort.
Naast de FISH-methode behoren tomografie (CT of MRI) tot de instrumentele diagnostische methoden voor het Angelman-syndroom. Deze methode helpt de toestand en de omvang van de hersenen te bepalen, en een elektro-encefalogram (EEG), dat laat zien hoe afzonderlijke delen van de hersenen functioneren.
Meestal stellen artsen de definitieve diagnose tussen de leeftijd van 3 en 7 jaar. Op dat moment vertonen de patiënten al de meeste symptomen en zijn de dynamieken van het ziekteverloop zichtbaar.
Welke tests zijn nodig?
Differentiële diagnose
Het Angelman-syndroom is een genetische aandoening die vrijwel geen specifieke manifestaties kent. De meeste symptomen kunnen zowel op AS als op andere genetische aandoeningen wijzen.
Differentiële diagnose van het Angelman-syndroom wordt uitgevoerd bij de volgende pathologieën:
- Pitt-Hopkins-syndroom (patiënten worden gekenmerkt door mentale retardatie, een vrolijk karakter, een glimlach, een vrij grote en brede mond, microcefalie wordt opgemerkt). Het verschil is dat ze in wakende toestand last hebben van hyperventilatieaanvallen en het inhouden van de adem.
- Syndroom van Christianson (patiënten zijn geestelijk gehandicapte personen met een vrolijke aard, niet in staat om te spreken, gekenmerkt door microcefalie, ataxie, convulsies en onwillekeurige spierbewegingen).
- Mowat-Wilsonsyndroom (symptomen: mentale retardatie, epileptische aanvallen, puntige kin, open mond, vrolijke gezichtsuitdrukking, microcefalie). Kenmerken: grote afstand tussen de ogen, schuin naar binnen gerichte ogen, ronde neuspunt, achterovergebogen oorschelp.
- Kabukisyndroom (gekenmerkt door lichte tot matige mentale retardatie, spraak- en motorische problemen, spierzwakte, epileptische aanvallen, microcefalie, lange tussenpozen tussen jeukaanvallen en verminderde coördinatie). Gekenmerkt door opgetrokken wenkbrauwen, een naar buiten gedraaide zijkant van het onderste ooglid, wijd uit elkaar staande ogen, lange ooglidspleten met lange, dikke wimpers.
- Rettsyndroom (differentiatie van AS bij vrouwen). Symptomen: vertraagde spraakontwikkeling, epileptische aanvallen, microcefalie. Het verschil is dat er geen vrolijke gezichtsuitdrukking is, er zijn aanvallen van apneu en apraxie, die in de loop van de tijd verergeren.
- Autosomaal recessief mentaal retardatiesyndroom 38 (symptomen: duidelijke mentale retardatie met vertragingen in motoriek en spraak, spierzwakte, voedingsproblemen in de kindertijd, impulsiviteit). Kenmerkend is de blauwe kleur van de iris.
- MECP 2-genduplicatiesyndroom (differentiatie van SA bij mannen). Symptomen: ernstige mentale retardatie, spierzwakte sinds de kindertijd, spraakproblemen of spraakgebrek, epilepsie. Onderscheidingen: progressieve myopathie, constant terugkerende infecties.
- Kleefstra-syndroom (symptomen: spraak- en denkproblemen, spierzwakte, slaapstoornissen, aandachtstekort, open mond, hyperactiviteit, toevallen, ataxie, evenwichtsstoornissen). Kenmerkende kenmerken: plat gezicht, korte, stompe neus, wijd uit elkaar staande ogen, grote, naar buiten staande onderlip, agressieve uitbarstingen.
- Smith-Magenis-syndroom (gekenmerkt door epileptische aanvallen, slaapproblemen, intellectuele en motorische ontwikkelingsstoornissen). Kenmerkende kenmerken zijn een breed en plat gezicht en een prominent voorhoofd.
- Syndroom van Koolen-de Vries (lichte tot matige verstandelijke beperking, spierzwakte, toevallen, vriendelijkheid). Kenmerkende kenmerken: lang gezicht met hoog voorhoofd, afstaande oren, scheve ogen, hoge gewrichtsmobiliteit, aangeboren hartafwijkingen.
- Syndroom van Phelan-McDermid (symptomen: mentale retardatie, spraakstoornissen of gebrek aan spraak). Kenmerken: grote handen met ontwikkelde spieren, spierzwakte vanaf de geboorte, zwak zweten.
Pathologieën zoals adenylsuccinaat-deficiëntie, autosomaal recessief mentaal retardatiesyndroom 1, chromosoom 2q23.1 duplicatiesyndroom, FOXG1-, STXBP1- of MEF2C-genhaploinsufficiëntiesyndromen en enkele andere kunnen “pronken” met symptomen die lijken op het Angelman-syndroom.
De taak van de arts is om een nauwkeurige diagnose te stellen, het Angelman-syndroom te onderscheiden van pathologieën met vergelijkbare symptomen, en een effectieve behandeling voor te schrijven die past bij het gediagnosticeerde stadium van de ziekte.
Met wie kun je contact opnemen?
Behandeling Angelman syndroom
Het Angelman-syndroom is een van die aandoeningen waarvoor de geneeskunde nog steeds op zoek is naar een effectieve behandeling. De etiologische behandeling van de ziekte bevindt zich in de ontwikkelingsfase met verschillende methoden en middelen, waarvan vele nog niet op mensen zijn getest. Dit betekent dat artsen zich voorlopig moeten beperken tot symptomatische therapie, wat op de een of andere manier helpt om de weinig benijdenswaardige situatie van kinderen en volwassenen met het marionetsyndroom, die lijden aan epileptische aanvallen, speekselvloed, hypotensie en slaapstoornissen, te verlichten.
Het is dus mogelijk om de frequentie en de ernst van epileptische aanvallen te verminderen met behulp van een goed gekozen anti-epilepticum. Maar het hele probleem is dat aanvallen bij patiënten met epilepsie zich onderscheiden van gewone epileptische aanvallen doordat ze worden gekenmerkt door meerdere soorten aanvallen, wat betekent dat de aandoening kan worden verlicht door meerdere medicijnen tegelijk toe te dienen.
De meest gebruikte anticonvulsiva voor de behandeling van het Angelman-syndroom zijn valproïnezuur, topiramaat, lamotrigine, levetiracetam, clonazepam en geneesmiddelen op basis daarvan. Minder vaak gebruikt zijn geneesmiddelen op basis van carmazepine, fenytoïne, fenobarbital en ethosuximide, omdat sommige hiervan een paradoxaal effect kunnen hebben, namelijk het versterken en de frequentie van epileptische aanvallen verhogen. Dit gebeurt wanneer het geneesmiddel als monotherapie wordt gebruikt.
Om speekselvloed te behandelen, worden meestal twee methoden gebruikt: medicinaal (medicijnen die de speekselproductie onderdrukken) en chirurgisch, waarbij de speekselklieren worden gereïmplanteerd. Maar in het geval van speekselvloed worden deze methoden als ineffectief beschouwd en blijft het probleem bestaan. Ouders en verzorgers van dergelijke patiënten moeten speciale aandacht aan dit probleem besteden, aangezien de patiënten zelf meestal geen controle hebben over hun speekselvloed en sommigen simpelweg niet in staat zijn om voor zichzelf te zorgen.
Een ander probleem is de korte slaapduur. Kinderen met het Angelman-syndroom slapen vaak niet meer dan 5 uur, wat een negatieve invloed heeft op de werking van het hele lichaam. Licht prikkelbare, actieve kinderen die dol zijn op spelletjes en communicatie (ook al beperken ze zich tot non-verbale uitingen) zijn overdag merkbaar moe. Om goed te kunnen uitrusten, heeft het lichaam een diepe, volle slaap nodig, maar daar zit nu juist de crux.
Het lijkt erop dat kalmerende middelen (fenothiazines en atypische antipsychotica) die het zenuwstelsel kalmeren, voldoende zouden moeten zijn om de slaap te verbeteren bij prikkelbare patiënten. Maar bij AS gaat het gebruik van dergelijke middelen gepaard met bijwerkingen. Daarom geven artsen nog steeds de voorkeur aan milde slaapmiddelen, zoals melatonine (een natuurlijk hormonaal middel op basis van het slaaphormoon), dat patiënten een uur voor het slapengaan innemen in de hoeveelheid van 1 tablet, en difenhydramine. De frequentie en dosering hiervan worden door de arts bepaald, afhankelijk van de toestand en leeftijd van de patiënt.
Soms hebben patiënten met het Angelman-syndroom problemen met de spijsvertering en de ontlasting. U kunt uw ontlasting verbeteren met laxeermiddelen (bij voorkeur kruiden).
Of je kunt het probleem anders benaderen, zoals Amerikaanse artsen deden, gebaseerd op bepaalde behandelmethoden voor autisme, omdat veel symptomen die kenmerkend zijn voor AS ook kenmerkend zijn voor autisme (impulsiviteit, onwillekeurige bewegingen, repetitieve handelingen, aandachtstekort, communicatieproblemen, enz.). Er werd opgemerkt dat de introductie van het hormoon secretine, dat de spijsvertering en de ontlasting normaliseert, een positief effect heeft op de aandacht van patiënten, en dat oxytocine helpt de cognitieve vaardigheden en het geheugen van het kind te verbeteren en gedrag te corrigeren.
Hormonen alleen zijn natuurlijk niet voldoende, zeker niet bij kinderen. Bij het Angelman-syndroom zijn gedragstherapie, samenwerking met een psycholoog en logopedist (die non-verbale communicatie en gebarentaal aanleren) aangewezen. De opvoeding van deze kinderen moet gebaseerd zijn op een individueel programma met medewerking van speciaal opgeleide leerkrachten, een psycholoog en ouders. Helaas is dit niet overal mogelijk en worden gezinnen met hun problemen aan hun lot overgelaten.
Omdat veel jonge patiënten met AS kampen met een lage spierspanning en gewrichtsproblemen, wordt er veel aandacht besteed aan fysiotherapie. Meestal maken artsen gebruik van paraffinebehandelingen, elektroforese en magneettherapie.
Actieve tonische massage en speciale oefeningen voor therapeutische fysieke training zullen het zieke kind helpen om na een tijdje weer op de been te komen en zelfverzekerd te lopen. Aquagym is hierbij bijzonder nuttig, en wordt aanbevolen voor SA in koud water. Het verhoogt de spierspanning en leert het kind zijn lichaam te beheersen en zijn bewegingen te coördineren.
Anticonvulsieve behandeling
Het gevaarlijkste symptoom van het Angelman-syndroom zijn aanvallen die lijken op epilepsie. Dit symptoom wordt bij 80% van de patiënten waargenomen, wat betekent dat ze allemaal effectieve anticonvulsiva voorgeschreven moeten krijgen.
Epileptische aanvallen worden behandeld met vitamines en anti-epileptica. Bij het Angelman-syndroom, dat gepaard gaat met een convulsiesyndroom, zijn vitamines van groep B, evenals vitamine C, D en E nuttig. Het is echter zeer gevaarlijk om in dit geval zelf vitaminetherapie voor te schrijven, omdat ongecontroleerde inname van vitamines de effectiviteit van anti-epileptica kan verminderen en nieuwe, ernstigere en langdurigere aanvallen kan veroorzaken.
De selectie van anti-epileptica en het voorschrijven van de effectieve dosering ervan moet ook door een specialist worden gedaan. Hij of zij beslist ook of één medicijn voldoende is of dat de patiënt gedurende langere tijd twee of meer medicijnen moet gebruiken.
Aan de meeste patiënten schrijven artsen medicijnen voor die valproïnezuur bevatten (Valproïnezuur, Depakine, Convulex, Valparin, enz.), die aanvallen voorkomen en de stemming en geestelijke gesteldheid van de patiënt verbeteren.
Valproïnezuur is verkrijgbaar in de vorm van tabletten, siroop en injectievloeistoffen. Het meest gebruikte medicijn is het geneesmiddel met verlengde afgifte "Depakine" in tabletten en als oplossing voor intraveneuze toediening. De dosering van het geneesmiddel wordt door de arts individueel bepaald, afhankelijk van het gewicht, de leeftijd en de conditie van de patiënt.
Het medicijn wordt 2 tot 3 keer per dag tijdens de maaltijd ingenomen. De gemiddelde dagelijkse dosis is 20-30 mg per kilogram lichaamsgewicht, met een maximum van 50 mg/kg per dag.
Contra-indicaties voor gebruik. Niet gebruiken bij lever- en pancreasfunctiestoornissen, hemorragische diathese, hepatitis, porfyrie en overgevoeligheid voor het geneesmiddel.
Bijwerkingen zijn onder meer trillende handen, spijsverterings- en stoelgangproblemen en veranderingen in lichaamsgewicht.
Topiramaat is ook een voorkeursgeneesmiddel voor SA. Het wordt geproduceerd in tabletvorm en wordt zowel als monotherapie als in combinatie met andere geneesmiddelen gebruikt.
Toedieningswijze en dosering. Neem de tabletten oraal in, ongeacht de voedselinname. De initiële dagelijkse dosis voor volwassenen is 25-50 mg, voor kinderen 0,5-1 mg/kg. Elke week wordt de dosering verhoogd volgens de instructies van de arts.
Het geneesmiddel mag niet worden gebruikt tijdens zwangerschap en borstvoeding, en ook niet bij overgevoeligheid voor de bestanddelen. Het geneesmiddel heeft veel verschillende bijwerkingen.
Medicijnen die een arts kan voorschrijven bij het Angelman-syndroom: Clomazepam, Rivotril, Lamotrigine, Seizar, Lamictal, Levetiracetam, Keppra, Epiterra, enz.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Traditionele geneeskunde en homeopathie
Traditionele geneeswijzen, zoals homeopathische preparaten, zijn uiteraard relatief veilig, maar de effectiviteit van een dergelijke behandeling voor het Angelman-syndroom is omstreden.
Hoewel traditionele therapieën nog steeds kunnen helpen, bijvoorbeeld bij het stoppen van epileptische aanvallen, kan een kruidenbehandeling in dit opzicht zeer effectief zijn.
Een medicinale collectie op basis van pioenroos, zoethout en kroos (de componenten worden in gelijke hoeveelheden ingenomen) biedt een goed effect. De kruiden moeten tot meel worden gemalen. Na twee weken na de start van de inname kunt u een aanzienlijke afname van de frequentie van aanvallen waarnemen.
Lavendelafkooksel (1 theelepel per glas kokend water) is ook effectief tegen krampen. Kook het mengsel 5 minuten en laat het een half uur trekken. Het medicijn wordt gedurende 14 dagen 's avonds ingenomen.
Een waterige (of alcoholische) infusie van hartgespan wordt als effectief beschouwd bij epileptische aanvallen.
Van de homeopathische preparaten ter voorkoming van aanvallen bij het Angelman-syndroom kunt u geneesmiddelen gebruiken op basis van kamille en hartgespan, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum en Arsenicum album. Houd er echter rekening mee dat alleen een homeopathisch arts in elk specifiek geval effectieve en veilige doses van geneesmiddelen kan voorschrijven.
Het voorkomen
Zoals de lezer waarschijnlijk al heeft begrepen, is de geneeskunde echter nog niet in staat om genmutaties en andere chromosomale afwijkingen te voorkomen en de situatie te corrigeren. Dit kan iedereen overkomen, omdat kinderen met het Angelman-syndroom geboren worden uit gezonde ouders, en genetica, momenteel een van de minst bestudeerde takken van de geneeskunde, dit nog niet kan verklaren.
Het enige wat we kunnen doen, is verantwoord omgaan met de zwangerschapsplanning, ons tijdig registreren en ons tijdig laten onderzoeken. Maar nogmaals, zo'n maatregel zal, net als alle onderzoeken, meer educatief dan preventief zijn. Maar jonge ouders weten van tevoren waar ze zich op moeten voorbereiden, en bij een positief antwoord zullen ze beslissen of ze een dergelijke verantwoordelijkheid als de opvoeding van een ziek kind op zich kunnen nemen.
Prognose
De prognose voor het Angelman-syndroom hangt af van de aard van de chromosomale afwijking en de tijdigheid van de detectie ervan. Kinderen bij wie chromosoom 15 "gaten" in de genen (deletie) bevat, worden het hardst getroffen. De kans dat deze patiënten weer kunnen lopen en praten is extreem klein. Andere gevallen kunnen worden gecorrigeerd met een zorgvuldige aanpak en liefde voor uw kind.
Helaas zullen dergelijke patiënten geen volwaardige leden van de maatschappij kunnen worden, ondanks het feit dat ze verre van dom zijn en spraak en de betekenis ervan begrijpen. Ze zullen echter de rest van hun leven problemen hebben met communicatie. Patiënten kunnen vanaf hun kindertijd gebarentaal leren, maar ze kunnen niet gedwongen worden om met woorden te communiceren. De woordenschat van "sprekende" patiënten is beperkt tot het minimum aan woorden dat ze in het dagelijks leven gebruiken (5-15 woorden).
Wat betreft de levensverwachting en de algemene gezondheid van patiënten met het Angelman-syndroom, schommelen de cijfers hier rond de gemiddelde waarden. Op volwassen leeftijd kampen patiënten meestal met gezondheidsproblemen zoals scoliose en obesitas, die met de juiste behandeling niet levensbedreigend zijn.