Medisch expert van het artikel

Nieuwe publicaties
Xeroderma pigmentosum
Laatst beoordeeld: 04.07.2025

Alle iLive-inhoud wordt medisch beoordeeld of gecontroleerd op feiten om zo veel mogelijk feitelijke nauwkeurigheid te waarborgen.
We hebben strikte richtlijnen voor sourcing en koppelen alleen aan gerenommeerde mediasites, academische onderzoeksinstellingen en, waar mogelijk, medisch getoetste onderzoeken. Merk op dat de nummers tussen haakjes ([1], [2], etc.) klikbare links naar deze studies zijn.
Als u van mening bent dat onze inhoud onjuist, verouderd of anderszins twijfelachtig is, selecteert u deze en drukt u op Ctrl + Enter.
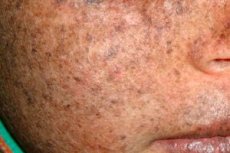
Xeroderma pigmentosum is een autosomaal recessieve genetische stoornis van DNA-herstel. De ziekte wordt veroorzaakt door mutaties in genen die betrokken zijn bij het herstel van DNA dat beschadigd is door ultraviolette straling en andere giftige stoffen.
Deze ziekte treft meestal kinderen, die ook wel 'kinderen van de nacht' worden genoemd. Veel voorkomende complicaties van de ziekte zijn basaalcelcarcinoom en andere kwaadaardige gezwellen van de huid, metastatisch maligne melanoom en plaveiselcelcarcinoom.
 [ 1 ]
[ 1 ]
Oorzaken xeroderma pigmentosum
Pathogenese
Een tekort aan UV-endonuclease-enzymen in de cellen van de patiënt (in fibroblasten) of de volledige afwezigheid ervan speelt een belangrijke rol bij het ontstaan van de ziekte. Dit enzym is verantwoordelijk voor de reproductie van DNA dat beschadigd is door ultraviolette straling. Volgens andere bronnen speelt een tekort aan het enzym DNA-polymerase-1 een belangrijke rol bij het ontstaan van de ziekte bij sommige patiënten. De ziekte ontwikkelt zich meestal onder invloed van straling met een golflengte van 280-310 nm. Men vermoedt dat pigmentxeroderma ontstaat door het binnendringen van verschillende fotodynamische stoffen in het lichaam of door een toename van porfyrines in de menselijke biologische omgeving.
In de vroege stadia van de ziekte worden hyperkeratose, atrofie van de epidermishaard, verdunning van de laag van Malpighia, een toename van melaninekorrels in de basale cellaag en chronische ontstekingsinfiltratie in de bovenste laag van de dermis (vooral rond de bloedvaten) waargenomen. Vervolgens worden degeneratieve veranderingen in collageen en elastische vezels waargenomen, en in het tumorstadium worden tekenen van huidkanker zichtbaar.
Symptomen xeroderma pigmentosum
Mannen en vrouwen worden even vaak door deze ziekte getroffen. De ziekte begint in de vroege kinderjaren, in de lente of zomer. De huid van de patiënt is bijzonder gevoelig voor zonlicht. Daarom verschijnt de eerste huiduitslag in de vorm van hyperpigmentatie, vergelijkbaar met een moedervlek, op delen van de huid die aan zonlicht worden blootgesteld. De uitslag neemt toe, het erytheem verergert, wordt donkerbruin, er verschijnen teleangiëctasieën, angiomen en atrofie. Vervolgens ontstaan papillomen en wratten (vooral op de huid van gezicht en hals), vlekken en zweren. Als gevolg van de littekenvorming van de zweren wordt de neus dunner ("vogelsnavel") en staat het ooglid naar buiten. Papillomen ontwikkelen zich meestal tot kwaadaardige tumoren. Pigmentxerodermie komt meestal voor in combinatie met spinocellulaire kanker, melanosarcomen.
Wat zit je dwars?
Stages
In het klinisch beloop van xeroderma pigmentosum worden vijf stadia onderscheiden: inflammatoir (erythemateus), hyperpigmentatie, atrofie, hyperkeratose en kwaadaardige tumoren.
Tijdens de ontstekingsfase (erythemateuze fase) verschijnen er zwellingen, rode vlekken en soms blaren en blaasjes op de huid die aan zonlicht worden blootgesteld (gezicht, nek, borst, armen, handen). Er is vrijwel geen huiduitslag op plekken die niet aan zonlicht worden blootgesteld.
Bij hyperpigmentatie ontstaan er lichtbruine, bruine hypergepigmenteerde vlekken die lijken op moedervlekken, in plaats van de rode vlekken.
Bij atrofie droogt de huid uit, wordt dunner en ontstaan er rimpels. Talrijke kleine, stervormige teleangiëctasieën en littekens met een glanzend oppervlak worden waargenomen op de huid van de lippen en neus. Door atrofie en littekens ontwikkelen zich microtomie (verkleining van de mondopening), ectropion, verdunning van de oren en neus, en atresie van de neusopening en de mond. Bij 80-85% van de patiënten worden de ogen aangetast: conjunctivitis, keratitis, schade aan het hoornvlies en de slijmvliezen, en verzwakking van het gezichtsvermogen worden opgemerkt. Dyschromie, teleangiëctasieën, hyperkeratotische veranderingen en tumoren worden waargenomen op de huid van de oogleden.
Bij hyperkeratose verschijnen wratachtige tumoren, papillomen, keratoacanthomen, fibromen, angiofibriomen en andere goedaardige tumoren in de hierboven beschreven pathologische haarden. Daarom rekenen sommige wetenschappers pigmentxerodermie tot de groep van precancereuze aandoeningen.
10-15 jaar na het begin van de ziekte verschijnen kwaadaardige huidtumoren (basalioom, endothelioom, angiosarcoom) in atrofisch veranderde haarden en pigmentvlekken. De tumoren vernietigen zich in korte tijd, zaaien uit naar inwendige organen en leiden tot de dood. In de inwendige organen en weefsels van sommige patiënten worden algemene dystrofische veranderingen waargenomen (syndactelia van de tweede en derde teen, tanddystrofie, volledige haaruitval, enz.).
Vormen
De neurologische vorm van xeroderma pigmentosum uit zich in twee syndromen.
Het Reed-syndroom wordt gekenmerkt door klinische manifestaties van xeroderma pigmentosum en microcefalie, idiopathie en een trage groei van het skelet. Bij de neurologische vorm van de ziekte is DNA-herstel moeilijk en leidt röntgentherapie tot een toename van de belangrijkste pathologische haarden.
Het De Sinctis-X Cocchione-syndroom wordt gekenmerkt door de volgende klinische symptomen:
- ontwikkeling van klinische tekenen van xeroderma pigmentosum op bijzonder lichtgevoelige huid;
- vroegtijdige verschijning van kwaadaardige tumoren;
- gelijktijdig beloop van spastische verlamming, microcefalie en dementie;
- aangeboren afwijkingen en dwerggroei;
- onderontwikkeling van de geslachtsklieren;
- frequente miskramen;
- recessieve overdracht door overerving.
Volgens sommige dermatologen is het Santis-Cacchione-syndroom geen op zichzelf staande ziekte, maar een ernstige en volledig uitgesproken klinische manifestatie van xeroderma pigmentosum.
Genetische vormen
Typen |
Gen |
Plaats |
Beschrijving |
Type A, I, XPA |
XPA |
9q22.3 |
Xeroderma pigmentosum (XP) groep A – klassieke vorm |
Type B, II, XPB |
XPB |
2q21 |
XP Groep B |
Type C, III, XPC |
XPC |
3p25 |
XP Groep C |
Type D, IV, XPD |
XPD ERCC6 |
19q13.2-q13.3, 10q11 |
XP Groep D of De Sanctis-Cacchione-syndroom |
Type E, V, XPE |
DDB2 |
11p12-p11 |
XP Groep E |
Type F, VI, XPF |
ERCC4 |
16p13.3-p13.13 |
XP Groep F |
Type G, VII, XPG |
RAD2ERCC5 |
13q33 |
XP groep G en COFS-syndroom (cerebro-oculo-facioskeletaal syndroom) type 3 |
Type V, XPV |
POLH |
6p21.1-p12 |
Variant Xeroderma Pigmentosa |
Wat moeten we onderzoeken?
Hoe te onderzoeken?
Met wie kun je contact opnemen?
Behandeling xeroderma pigmentosum
Antimalariamiddelen (Delagyl, Plaquenil, Resoquin, enz.) beschermen het DNA tegen ultraviolette straling, remmen het depolymerisatieproces en verminderen de gevoeligheid (fotosensibilisatie) van de huid voor zonlicht. Deze middelen hebben ontstekingsremmende en hyposensibiliserende eigenschappen. Het wordt aanbevolen om een algemene behandeling te combineren met vitaminetherapie (B1, B2, PP, B6, B12, A, E), antihistaminica (Tavegil, difenhydramine, Suprastin) en desensibiliserende middelen (natriumthiosulfaat, 10% calciumchloride, intraveneus 10 ml).
Voor plaatselijke behandeling worden zonnebrandcrèmes en -zalven gebruikt.
Bij de tumorvorm van pigmentxeroderma worden chirurgische methoden, vloeibare stikstof en laserstralen gebruikt. Draag losse kleding, een zonnehoed en handschoenen om uzelf tegen de zon te beschermen.
Prognose
De meeste patiënten (2/3) sterven voordat ze de leeftijd van 15 jaar bereiken. Sommige patiënten kunnen 40-50 jaar oud worden.
[ 20 ]