Prionen - veroorzakers van prionziekten
Last reviewed: 01.06.2018

We hanteren strikte richtlijnen voor bronnen en linken alleen naar gerenommeerde medische websites, academische onderzoeksinstellingen en, waar mogelijk, medisch peer-reviewed onderzoek. De nummers tussen haakjes ([1], [2], enz.) zijn klikbare links naar deze onderzoeken.
Als u van mening bent dat onze content onjuist, verouderd of anderszins twijfelachtig is, selecteer deze dan en druk op Ctrl + Enter.
Langzame virusinfecties worden gekenmerkt door speciale criteria:
- een ongewoon lange incubatietijd (maanden, jaren);
- een specifieke laesie van organen en weefsels, voornamelijk het centrale zenuwstelsel;
- langzame, gestage progressie van de ziekte;
- onvermijdelijke fatale afloop.

Sommige ziekteverwekkers die acute virusinfecties veroorzaken, kunnen ook langzame virusinfecties veroorzaken. Zo veroorzaakt het mazelenvirus soms SSPE, en het rubellavirus progressieve congenitale rubella en rubella panencefalitis.
Een typische langzame virusinfectie bij dieren wordt veroorzaakt door het visna/madi-virus, een retrovirus. Het is de veroorzaker van een langzame virusinfectie en progressieve longontsteking bij schapen. De witte hersenstof wordt vernietigd, er ontstaat verlamming (visna - wegkwijnen); er treedt chronische ontsteking van de longen en milt op.
Ziekten die qua kenmerken vergelijkbaar zijn met langzame virusinfecties, worden veroorzaakt door prionen – de verwekkers van prioninfecties. Prionziekten zijn een groep progressieve aandoeningen van het centrale zenuwstelsel bij mens en dier. Bij mensen is de functie van het centrale zenuwstelsel verstoord, treden persoonlijkheidsveranderingen en bewegingsstoornissen op. De symptomen van de ziekte duren meestal enkele maanden tot enkele jaren en leiden tot de dood. Voorheen werden prioninfecties beschouwd als één van de zogenaamde verwekkers van langzame virusinfecties.
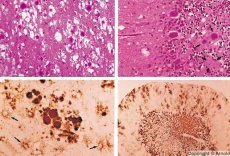
Sommige verwekkers van prionziekten hopen zich eerst op in lymfeweefsel. Prionen die de hersenen binnendringen, hopen zich in grote hoeveelheden op en veroorzaken amyloïdose (extracellulaire dysproteïnose, gekenmerkt door de afzetting van amyloïde met de ontwikkeling van atrofie en sclerose van het weefsel) en astrocytose (proliferatie van astrocytaire neuroglia, hyperproductie van gliavezels). Fibrillen, aggregaten van eiwitten of amyloïde en spongiforme veranderingen in de hersenen (overdraagbare spongiforme encefalopathieën) worden gevormd. Dit leidt tot gedragsveranderingen, een verstoorde bewegingscoördinatie en uitputting met fatale afloop. Er wordt geen immuniteit opgebouwd. Prionziekten zijn conformatieziekten die ontstaan als gevolg van een onjuiste vouwing (schending van de correcte conformatie) van cellulaire eiwitten die nodig zijn voor de normale werking van het lichaam. De routes van priontransmissie zijn divers:
- via het voedsel - geïnfecteerde producten van dierlijke oorsprong, voedseladditieven van rauwe runderorganen, enz.:
- overdracht via bloedtransfusie, toediening van geneesmiddelen van dierlijke oorsprong, orgaan- en weefseltransplantatie, gebruik van geïnfecteerde chirurgische en tandheelkundige instrumenten;
- overdracht via immunobiologische preparaten (besmetting van 1500 schapen met PrP''' door hersenformolvaccin van zieke schapen is bekend).
Pathologische prionen die de darm zijn binnengedrongen, worden naar het bloed en de lymfe getransporteerd. Na perifere replicatie in de milt, blindedarm, amandelen en andere lymfoïde weefsels worden ze via de perifere zenuwen naar de hersenen getransporteerd (neuro-invasie). Directe penetratie van prionen in de hersenen via de bloed-hersenbarrière is mogelijk. Voorheen werd gedacht dat het centrale zenuwstelsel het enige weefsel is waar pathologische prionen zich ophopen, maar er zijn studies verschenen die deze hypothese hebben veranderd. Het bleek dat de ophoping van prionen in de milt verband houdt met de toename en het functioneren van folliculaire dendritische cellen.
 [
[ Eigenschappen van prionen
De normale cellulaire isovorm van het prioneiwit met een molecuulgewicht van 33-35 kDa wordt bepaald door het prioneiwitgen (het priongen - PrNP) bevindt zich op het 20e menselijke chromosoom. Het normale gen bevindt zich op het celoppervlak (verankerd in het membraan door het glycoproteïne van het molecuul) en is gevoelig voor protease. Het reguleert de overdracht van zenuwimpulsen, dagelijkse cycli, oxidatieprocessen, neemt deel aan het kopermetabolisme in het centrale zenuwstelsel en aan de regulatie van de deling van beenmergstamcellen. Daarnaast wordt het priongen aangetroffen in de milt, lymfeklieren, huid, maag-darmkanaal en folliculaire dendritische cellen.
Proliferatie van pathologische prionen
De transformatie van prionen naar veranderde vormen vindt plaats wanneer het kinetisch gecontroleerde evenwicht tussen hen verstoord raakt. Dit proces wordt versterkt door een toename van de hoeveelheid pathologisch (PrP) of exogeen prion. PrP is een normaal eiwit dat verankerd is in het celmembraan. PrP' is een bolvormig hydrofoob eiwit dat aggregaten vormt met zichzelf en PrP'' op het celoppervlak: hierdoor wordt PrP' omgezet in PrP'' en gaat de cyclus verder. De pathologische vorm van PrP''' hoopt zich op in neuronen, waardoor de cel een sponsachtig uiterlijk krijgt.
Kuru
Prionziekte, voorheen veel voorkomend onder de Papoea's (wat trillen of schudden betekent) in het oostelijke deel van het eiland Nieuw-Guinea. De besmettelijke eigenschappen van de ziekte werden bewezen door K. Gajdusek. De ziekteverwekker wordt overgedragen via voedsel als gevolg van ritueel kannibalisme - het eten van de onvoldoende gekookte, met prionen geïnfecteerde hersenen van overleden familieleden. Door schade aan het centrale zenuwstelsel worden beweging en loopvermogen verstoord, treden koude rillingen en euforie ("lachendood") op. De incubatietijd duurt 5-30 jaar. De patiënt overlijdt na een jaar.
De ziekte van Creutzfeldt-Jakob
Prionziekte, die zich manifesteert als dementie, visuele en cerebellaire stoornissen en bewegingsstoornissen met een fatale afloop na 4-5 maanden ziekte bij de klassieke variant van de ziekte van Creutzfeldt-Jakob en na (3-14 maanden bij de nieuwe variant van de ziekte van Creutzfeldt-Jakob). De incubatietijd kan oplopen tot 20 jaar. Verschillende infectieroutes en oorzaken van de ziekte zijn mogelijk:
- bij consumptie van dierlijke producten die onvoldoende zijn verhit, zoals vlees en hersenen van koeien met boviene spongiforme encefalopathie;
- bij weefseltransplantatie, zoals hoornvliestransplantatie, bloedtransfusie, gebruik van hormonen en andere biologisch actieve stoffen van dierlijke oorsprong, gebruik van catgut, besmette of onvoldoende gesteriliseerde chirurgische instrumenten, prosectorale manipulaties;
- bij hyperproductie van PrR en andere omstandigheden die het proces van de omzetting van PrR' in PrR stimuleren".
De ziekte kan ook ontstaan als gevolg van een mutatie of insertie in het priongengebied. Een familiaire aard van de ziekte komt vaak voor vanwege de genetische aanleg voor de ziekte van Creutzfeldt-Jakob. Bij de nieuwe variant van de ziekte van Creutzfeldt-Jakob ontwikkelen de aandoeningen zich op jongere leeftijd (gemiddelde leeftijd 28 jaar), in tegenstelling tot de klassieke variant (gemiddelde leeftijd 65 jaar). Bij de nieuwe variant van de ziekte van Creutzfeldt-Jakob hoopt abnormaal prioneiwit zich niet alleen op in het centrale zenuwstelsel, maar ook in lymforeticulair weefsel, waaronder de amandelen.
Gerstmann-Sträussler-Scheinker-syndroom
Erfelijke prionziekte, gepaard gaand met dementie, hypotonie, slikproblemen (dysfagie), dysartrie. Komt vaak voor in de familie. De incubatietijd is 5 tot 30 jaar. De ziekte treedt op tussen de 50 en 60 jaar en de duur varieert van 5 tot 13 jaar.
Erfelijke dodelijke slapeloosheid
Een auto-immuunziekte met progressieve slapeloosheid, sympathische hyperreactiviteit (hypertensie, hyperthermie, hyperhidrose, tachycardie), tremor, ataxie, multiklonen en hallucinaties. De slaap is ernstig verstoord. De dood treedt op bij progressie van cardiovasculair falen.
Schrapen
Scrapie (van het Engelse scrape - schrapen) is een prionziekte bij schapen en geiten (scabiës), die gepaard gaat met schade aan het centrale zenuwstelsel, progressieve bewegingsstoornissen, hevige jeuk aan de huid (scabiës) en eindigt met de dood van het dier.
Boviene spongiforme encefalopathie
Een runderziekte die gekenmerkt wordt door schade aan het centrale zenuwstelsel, verminderde bewegingscoördinatie en de onvermijdelijke dood van het dier. De epidemie brak voor het eerst uit in Groot-Brittannië. De ziekte werd veroorzaakt door het voeren van dieren met vlees- en beendermeel dat pathologische prionen bevatte. De incubatietijd varieert van 1,5 tot 15 jaar. De hersenen, het ruggenmerg en de oogbollen van dieren raken het vaakst geïnfecteerd.
Laboratoriumdiagnostiek van prionziekten
Tijdens de diagnostiek worden spongiforme veranderingen in de hersenen, astrocytose (gliose) en de afwezigheid van ontstekingsinfiltraten opgemerkt. De hersenen worden gekleurd op amyloïde. Eiwitmarkers van prionische hersenaandoeningen worden in de cerebrospinale vloeistof (CSF) gedetecteerd (met behulp van ELISA). Er wordt een genetische analyse van het priongen (PCR) uitgevoerd.
Preventie van prionziekten
Autoclaveren (bij 134 °C gedurende 18 minuten; bij 121 °C gedurende 1 uur), verbranding, aanvullende behandeling met bleekmiddel en een 1-normale NaCl-oplossing gedurende 1 uur worden aanbevolen voor de decontaminatie van instrumenten en omgevingsobjecten. Voor niet-specifieke profylaxe zijn beperkingen ingevoerd op het gebruik van geneesmiddelen van dierlijke oorsprong en is de productie van hypofysehormonen van dierlijke oorsprong verboden. Transplantatie van de dura mater is beperkt. Rubberen handschoenen worden gebruikt bij het werken met de weefselvloeistoffen van patiënten.