Medisch expert van het artikel

Nieuwe publicaties
Ushersyndroom
Laatst beoordeeld: 04.07.2025

Alle iLive-inhoud wordt medisch beoordeeld of gecontroleerd op feiten om zo veel mogelijk feitelijke nauwkeurigheid te waarborgen.
We hebben strikte richtlijnen voor sourcing en koppelen alleen aan gerenommeerde mediasites, academische onderzoeksinstellingen en, waar mogelijk, medisch getoetste onderzoeken. Merk op dat de nummers tussen haakjes ([1], [2], etc.) klikbare links naar deze studies zijn.
Als u van mening bent dat onze inhoud onjuist, verouderd of anderszins twijfelachtig is, selecteert u deze en drukt u op Ctrl + Enter.
Het syndroom van Usher is een erfelijke aandoening die zich manifesteert als volledige doofheid vanaf de geboorte, evenals progressieve blindheid met de leeftijd. Het verlies van gezichtsvermogen gaat gepaard met retinitis pigmentosa, een proces van pigmentdegeneratie van het netvlies. Veel mensen met het syndroom van Usher hebben ook ernstige evenwichtsproblemen.
Epidemiologie
Dankzij het onderzoek kon worden vastgesteld dat het syndroom van Usher ongeveer 8% van de onderzochte doofstomme kinderen treft (de tests werden uitgevoerd in speciale instellingen voor doofstommen). Pigmentaire retinitis werd waargenomen bij 6-10% van de patiënten met aangeboren doofheid, wat op zijn beurt wordt waargenomen bij ongeveer 30% van de mensen met een pigmentaire retinale aandoening.
Men denkt dat deze ziekte zich wereldwijd bij ongeveer 3-10 op de 100.000 mensen manifesteert. Het komt even vaak voor bij vrouwen als bij mannen. Ongeveer 5-6% van de wereldbevolking lijdt aan dit syndroom. Ongeveer 10% van alle gevallen van ernstige doofheid bij kinderen wordt veroorzaakt door het syndroom van Usher type I, en type II.
In de Verenigde Staten zijn type 1 en 2 de meest voorkomende typen. Samen zijn ze verantwoordelijk voor ongeveer 90 tot 95 procent van alle gevallen van het Ushersyndroom bij kinderen.
Oorzaken Ushersyndroom
Het syndroom van Usher type I, II en III heeft een autosomaal recessieve oorzaak, terwijl type IV wordt beschouwd als een X-chromosoomafwijking. De oorzaken van blindheid en doofheid die bij dit syndroom voorkomen, zijn nog niet voldoende onderzocht. Er wordt aangenomen dat mensen met deze ziekte overgevoelig zijn voor componenten die de DNA-structuur kunnen beschadigen. Daarnaast kan deze ziekte gepaard gaan met aandoeningen van het immuunsysteem, maar in dit geval is er geen exact beeld van dit proces.
In 1989 werden voor het eerst chromosomale afwijkingen vastgesteld bij patiënten met type 2-ziekte, wat in de toekomst mogelijk kan leiden tot een manier om de genen die het syndroom veroorzaken te isoleren. Mogelijk kunnen deze genen ook bij dragers worden geïdentificeerd en kunnen er speciale prenatale genetische tests worden ontwikkeld.
 [ 8 ]
[ 8 ]
Risicofactoren
Het syndroom wordt overgeërfd wanneer beide ouders de ziekte hebben, d.w.z. het wordt overgeërfd via een recessief type. Een kind kan de ziekte ook erven als zijn ouders drager zijn van het gen. Als beide toekomstige ouders dit gen hebben, is de kans op een kind met dit syndroom 1 op 4. Iemand die slechts één gen voor het syndroom heeft, wordt beschouwd als drager, maar heeft geen symptomen van de aandoening. Tegenwoordig is het nog niet mogelijk om vast te stellen of iemand het gen voor deze ziekte heeft.
Wanneer een kind geboren wordt uit ouders waarvan er één dit gen niet heeft, is de kans dat hij het syndroom erft erg klein, maar hij zal zeker drager zijn.
Symptomen Ushersyndroom
Symptomen van het syndroom van Usher zijn onder meer gehoorverlies en een abnormale ophoping van pigmentcellen in de oogstructuren. De patiënt ontwikkelt vervolgens degeneratie van het netvlies, wat leidt tot verslechtering van het gezichtsvermogen en in de ernstigste gevallen uiteindelijk tot gezichtsverlies.
Perceptief gehoorverlies kan mild of volledig zijn en ontwikkelt zich meestal niet vanaf de geboorte. Netvliespigmentziekte kan zich echter al in de kindertijd of later ontwikkelen. Testresultaten hebben aangetoond dat de centrale gezichtsscherpte jarenlang behouden kan blijven, zelfs wanneer het perifere zicht achteruitgaat (een aandoening die 'tunnelvisie' wordt genoemd).
Dit zijn de voornaamste verschijnselen van de ziekte, die soms gepaard gaan met andere aandoeningen, zoals psychoses en andere psychische stoornissen, problemen met het binnenoor en/of staar.
Vormen
Tijdens het onderzoek werden drie typen van de ziekte geïdentificeerd en een vierde vorm, die vrij zeldzaam is.
Type I van de ziekte wordt gekenmerkt door aangeboren volledige doofheid en evenwichtsstoornissen. Vaak beginnen deze kinderen pas rond de leeftijd van 1,5 jaar te lopen. De achteruitgang van het gezichtsvermogen begint meestal rond de leeftijd van 10 jaar en de uiteindelijke ontwikkeling van nachtblindheid begint rond de leeftijd van 20 jaar. Kinderen met dit type ziekte kunnen een progressieve achteruitgang van het perifere zicht ontwikkelen.
Bij type II wordt matige of aangeboren doofheid waargenomen. In dit geval treedt er vaak geen verslechtering van de gedeeltelijke doofheid meer op. Pigmentaire retinitis begint zich te ontwikkelen rond het einde van de adolescentie of na 20 jaar. Nachtblindheid begint meestal rond de leeftijd van 29-31 jaar. De visuele scherptevermindering bij type II pathologie verloopt over het algemeen iets langzamer dan bij type I.
Type III van de ziekte wordt gekenmerkt door progressief gehoorverlies, dat gewoonlijk begint tijdens de puberteit. In dezelfde periode (iets later dan het gehoorverlies) ontwikkelt zich geleidelijk retinitis pigmentosa, wat een factor kan worden in de ontwikkeling van progressieve blindheid.
Manifestaties van type IV-pathologie komen voornamelijk voor bij mannen. In dit geval worden ook progressieve aandoeningen en gehoor- en gezichtsverlies waargenomen. Deze vorm is zeer zeldzaam en heeft meestal een X-chromosomaal karakter.
Diagnostics Ushersyndroom
De diagnose van het syndroom van Usher wordt gesteld op basis van de combinatie van plotselinge doofheid en progressief verlies van het gezichtsvermogen bij de patiënt.
Testen
Er kan een speciale genetische test worden voorgeschreven om de mutatie op te sporen.
Er zijn elf genetische loci gevonden die de ontwikkeling van het syndroom van Usher kunnen veroorzaken, en negen genen zijn geïdentificeerd die met zekerheid de oorzaak zijn van de stoornis:
- Type 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Type 2: ush2a, VLGR1, WHRN.
- Syndroom van Usher type 3: USH3A.
Wetenschappers van het NIDCD hebben samen met collega's van universiteiten in New York en Israël een mutatie genaamd R245X in het Pcdh15-gen geïdentificeerd. Deze mutatie is verantwoordelijk voor een groot percentage van de gevallen van Ushersyndroom type 1 bij de Joodse bevolking.
Voor meer informatie over laboratoria die klinische proeven uitvoeren, gaat u naar https://www.genetests.org en zoekt u in de labgids naar 'Ushersyndroom'.
Voor meer informatie over bestaande klinische onderzoeken met genetische tests voor het syndroom van Usher gaat u naar https://www.clinicaltrials.gov en zoekt u naar "Ushersyndroom" of "genetische tests voor het syndroom van Usher".
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Instrumentele diagnostiek
Er bestaan verschillende methoden voor instrumentele diagnostiek:
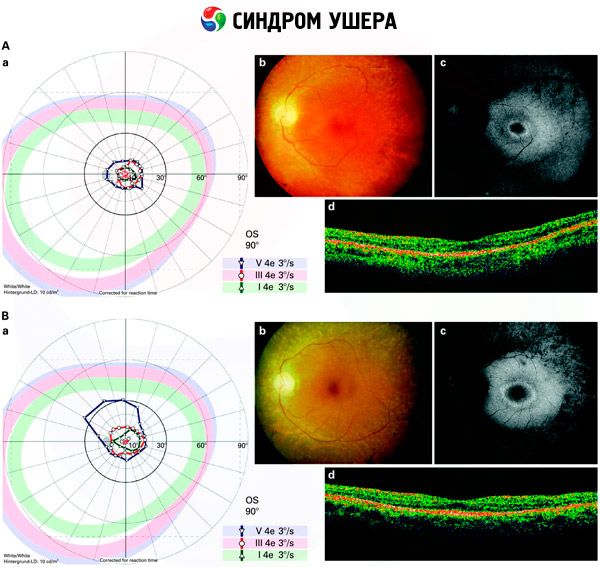
- Onderzoek van het fundus om de aanwezigheid van pigmentvlekken op het netvlies op te sporen, evenals vernauwingen van de vaten in het netvlies;
- Elektroretinografie, waarmee initiële degeneratieve afwijkingen in het netvlies van het oog kunnen worden gedetecteerd. Het toont de uitdoving van elektroradiografische paden;
- Bij een elektronystagmogram (ENG) worden onwillekeurige oogbewegingen gemeten, die kunnen wijzen op de aanwezigheid van een onevenwicht.
- Audiometrie, waarmee de aanwezigheid van doofheid en de ernst ervan worden vastgesteld.
Differentiële diagnose
Het syndroom van Usher moet worden onderscheiden van vergelijkbare aandoeningen.
Het syndroom van Hallgren, dat wordt gekenmerkt door aangeboren gehoorverlies en progressief verlies van het gezichtsvermogen (ook cataract en nystagmus ontwikkelen zich). Andere symptomen zijn ataxie, psychomotorische stoornissen, psychose en mentale retardatie.
Het syndroom van Alstrom is een erfelijke ziekte waarbij het netvlies degenereert, wat leidt tot verlies van het centrale zicht. Dit syndroom gaat gepaard met obesitas bij kinderen. Tegelijkertijd beginnen diabetes mellitus en gehoorverlies zich na 10 jaar te ontwikkelen.
Rodehond bij een zwangere vrouw in het eerste trimester kan verschillende afwijkingen in de ontwikkeling van het kind veroorzaken. Gevolgen van een dergelijke afwijking zijn onder meer gehoorverlies, problemen met het gezichtsvermogen en daarnaast diverse ontwikkelingsstoornissen.
Met wie kun je contact opnemen?
Behandeling Ushersyndroom
Er is momenteel geen genezing mogelijk voor het syndroom van Usher. Daarom bestaat de behandeling in dit geval voornamelijk uit het vertragen van het proces van gezichtsverlies en het compenseren van gehoorverlies. Mogelijke behandelmethoden zijn onder andere:
- Het nemen van vitamine A (sommige oogartsen geloven dat hoge doses vitamine A-palmitaat de progressie van retinitis pigmentosa kunnen vertragen, maar niet stoppen);
- Implantatie van speciale elektronische apparaten in de oren van de patiënt (hoortoestellen, cochleaire implantaten).
Oogartsen adviseren dat de meeste volwassenen met veel voorkomende vormen van retinitis pigmentosa dagelijks 15.000 IE (internationale eenheden) vitamine A-palmitaat innemen onder toezicht. Omdat mensen met het Ushersyndroom type 1 niet in het onderzoek zijn opgenomen, worden hoge doses vitamine A voor deze groep patiënten niet aanbevolen. Mensen die overwegen vitamine A te gebruiken, dienen deze behandelingsoptie met hun arts te bespreken. Andere aanbevelingen voor deze behandelingsoptie zijn:
- Verander uw dieet en eet voedingsmiddelen die veel vitamine A bevatten.
- Vrouwen die zwanger willen worden, moeten drie maanden voordat ze zwanger willen worden stoppen met het nemen van hoge doseringen vitamine A vanwege een verhoogd risico op geboorteafwijkingen.
- Vrouwen die zwanger zijn, moeten stoppen met het nemen van hoge doseringen vitamine A vanwege een verhoogd risico op geboorteafwijkingen.
Het is ook belangrijk om zo'n kind te laten wennen aan het sociale leven. Hiervoor is de hulp nodig van leerkrachten speciaal onderwijs en psychologen. Indien de patiënt progressief gezichtsverlies begint te ervaren, moet hij gebarentaal leren gebruiken.
Prognose
Het syndroom van Usher heeft een ongunstige prognose. Het gezichtsveld en de gezichtsscherpte beginnen bij de meeste patiënten met deze ziekte, ongeacht het type, binnen 20-30 jaar af te nemen. In sommige gevallen treedt volledig bilateraal gezichtsverlies op. Gehoorverlies, dat altijd gepaard gaat met doofheid, ontwikkelt zich zeer snel tot volledig bilateraal gehoorverlies.