Medisch expert van het artikel

Nieuwe publicaties
Hamartoom
Laatst beoordeeld: 07.06.2024

Alle iLive-inhoud wordt medisch beoordeeld of gecontroleerd op feiten om zo veel mogelijk feitelijke nauwkeurigheid te waarborgen.
We hebben strikte richtlijnen voor sourcing en koppelen alleen aan gerenommeerde mediasites, academische onderzoeksinstellingen en, waar mogelijk, medisch getoetste onderzoeken. Merk op dat de nummers tussen haakjes ([1], [2], etc.) klikbare links naar deze studies zijn.
Als u van mening bent dat onze inhoud onjuist, verouderd of anderszins twijfelachtig is, selecteert u deze en drukt u op Ctrl + Enter.
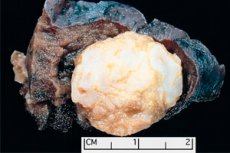
Tumor-achtige vorming gelokaliseerd in elk anatomisch gebied als gevolg van abnormale groei van goedaardig weefsel, in de geneeskunde wordt gedefinieerd als hamartoma (van Griekse hamartia - fout, defect). [1] ]
Epidemiologie
Statistisch gezien zijn Hamartoma's goed voor 1,2% van goedaardige neoplasmata. De prevalentie van longhamartomen wordt geschat op ongeveer 0,25% van de algemene bevolking en is goed voor maximaal 8% van alle pulmonale neoplasmata. De meeste longhamartomen worden overigens gediagnosticeerd bij patiënten van 40 tot 70 jaar, maar zijn zeer zeldzaam in de pediatrische praktijk.
Over het algemeen worden de meeste hamartomen gediagnosticeerd bij mannen, hoewel ze in de nier vaker voorkomen bij vrouwen en worden geïdentificeerd op middelbare leeftijd.
Ongeveer 5% van de goedaardige borsttumoren zijn hamartomen, en ze treffen meestal vrouwen ouder dan 35 jaar.
80-90% van de hamartomateuze laesies van de hersenen en meer dan 50% hamartomen van het hart worden geassocieerd met tubereuze sclerose.
Oorzaken Hamartomen
Gamartoma's behoren tot aangeboren misvormingen en zijn formaties van goedaardige karakter, die worden gevormd uit mesenchymale weefsels afkomstig van kiemplaten. En de oorzaken van hun optreden worden geassocieerd met ongecontroleerde celdeling van cytologisch normale weefsels (bind, gladde spieren, vet of kraakbeen), kenmerkend voor een gegeven anatomische locatie, en hun focale overgroei tijdens embryogenese van bijna elke orgaan- of anatomische structuur.
Het optreden van meerdere hamartomen bij dezelfde patiënt wordt vaak hamartomatose of pleiotrope hamartoom genoemd.
Deze tumoren kunnen sporadisch optreden of in aanwezigheid van bepaalde autosomale dominante erfelijke ziekten evenals genetisch bepaalde syndromen.
In veel gevallen vormen hamartomen wanneer een zeldzame genetische ziekte van multisystemische aard - tubereuze sclerose -manifesteert kort na de geboorte, of in de familiale ziekte van Reckinghausen - neurofibromatose type 1. [2]
Risicofactoren
De belangrijkste risicofactoren voor de vorming van hamartoma zijn onder meer de aanwezigheid van zogenaamde genetische syndromen van hamartomateuze polyposis in de geschiedenis van de patiënten, waaronder:
- Multiple hamartoma-syndroom- Cowden-syndroom, waarbij meerdere hamartomen van ecto-, ento- en mesodermale oorsprong worden gevormd, gastro-intestinale polyposis en mucocutane manifestaties worden waargenomen;
- Peutz-Jeghers-Turen syndroom (gekenmerkt door de ontwikkeling van goedaardige hamartomateuze poliepen in het maagdarmkanaal);
- Proteus Syndrome;
- Weil Syndrome
- Bannayan-Riley-Ruvalcaba-syndroom, dat, net als het Cowden-syndroom, meerdere hamartomen (hamartomateuze poliepen) van de darm produceert;
- Carney-Stratakis Syndrome en Carney Complex.
Bovendien vormen hamartomen bij patiënten met erfelijk Watson-syndroom en in gevallen van sporadisch voorkomende of aangeboren Pallister-Hall-syndroom met hypothalamisch hamartoma en polydactyly.
Pathogenese
Het mechanisme van verhoogde proliferatie van germinale weefsels met de vorming van tumorachtige misvormingen in verschillende organen wordt verklaard door chromosomale afwijkingen en genmutaties die spontaan of geërfd kunnen worden.
Bij tubereuze sclerose zijn mutaties in de TSC1- of TSC2-genen - tumorsuppressoren die overmatige proliferatie voorkomen en remmen - te snelle of ongecontroleerde celgroei en-afdeling - geïdentificeerd. En bij neurofibromatosis type 1 en Watson-syndroom - kiemlijnmutaties van het mitochondriale tumorsuppressorgen NF1.
In hamartoma-tumorsyndroom, dat Cowden, Protea, Bannayan-Riley-Ruvalcaba en juveniele polyposis syndromen combineert, wordt pathogenese geassocieerd met mutatie van het PTEN-gen, dat codeert voor een enzym dat betrokken is bij de regulatie van proliferatie en wordt beschouwd als een tumor-onderdrukken.
Mutaties in het STK11-gen dat codeert voor de structuur en functie van een van de transmembraanserine-enzymen, die het vermogen om de celdeling te beperken, leiden tot het Peutz-Jeghers-Turen-syndroom, met de ontwikkeling van darmpoliepen en gepigmenteerde huidletsels. Een mutatie in het Gli3-gen, een transcriptiefactor die betrokken is bij de vorming van intra-uteriene weefsel, is geïdentificeerd in het Pallister-Hall-syndroom.
Aldus leidt ongecontroleerde celgroei als gevolg van genmutaties tot hamartoomvorming.
Symptomen Hamartomen
Afhankelijk van de lokalisatie van hamartomen, worden hun typen onderscheiden en heeft elk van hen zijn eigen structuur en symptomatologie.
Hamartoma van de long
Longhamartoom kan zich vormen in alle lob en perifere delen van de longen en bestaat uit normale weefsels aanwezig in de longen: vet, epitheliaal, vezelachtig en kraakbeenachtig. In 80% van de gevallen overheerst de chondroid-component (hyaline kraakbeencellen) met de opname van adipocyten - vetweefselcellen en epitheelcellen van de luchtwegen. [3]
De eerdere namen: Chondroid-hamartoma, mesenchymoma, chondromateuze hamartoma of hamartochondroma worden momenteel niet aanbevolen door WHO.
Mesenchymaal cystisch hamartoma van de long is daarentegen minder gebruikelijk en wordt bij de meeste patiënten geassocieerd met het Cowden-syndroom.
Hamartomateuze laesie van de long manifesteert zich misschien niet, maar kan symptomen veroorzaken in de vorm van chronische hoest (vaak met hemoptysis), piepend bij het ademen en ademhalingsmoeilijkheden. [4]
Een hamartoma van het hart
Goedaardige primaire cardiale tumoren bij volwassenen omvatten volwassen myocytenhamartoom, en bij zuigelingen en kinderen met tubereuze sclerose, rabdomyoma, dat wil zeggen myocardiale hamartoma van de ventrikels of interventriculair septum. [5] ]
Rijpe cardiomyocytenhamartoma ontwikkelt zich in de ventriculaire wand (en zelden in de atria) en kan verschijnen als meerdere laesies die dichte massa's zijn die nauw verbonden zijn met het onderliggende myocardium. De tumor kan symptomen van hartfalen veroorzaken: pijn op de borst, hartkloppingen en aritmieën, hartgerurft, oedeem, dyspneu, cyanose.
Cardiale rabdomyomen, waarvan de meeste binnen het eerste levensjaar worden gediagnosticeerd, zijn samengesteld uit hartspierweefsel gevormd door embryonale myoblasten en hebben het uiterlijk van vaste focale massa's zonder capsule.
Meestal presenteren deze hamartomen asymptomatisch en regresseren vóór de leeftijd van 4 jaar.
Hamartomateuze laesies worden door sommige experts ook beschouwd als geassocieerd met het Carney-complex myxoma van het hart. [6]
Gamartoma van het maagdarmkanaal
Maaghamartoma is een mesenchymale massa in de vorm van een epitheliale hyperplastische poliep van de maag, Peutz-Jeghers-poliep en een zeldzame myoepitheliale hamartoma-met hypertrofische gladde spierbundels. Andere namen voor dit hamartoma zijn Myoglandular Hamartoma, adenomyomatous hamartoma en maagadenomyoom. Typische klinische manifestaties omvatten dyspepsie, epigastrische pijn en bovenste GI-bloedingen. [7], [8]
Meer informatie in het materiaal maagpolyposis
Een darmhamartoma is een hamartomateuze of hyperplastische poliep van de dikke darm, gediagnosticeerd als een adenomateus of buisvormig adenoom. Wanneer het hamartoma is gelokaliseerd in de klier van de brunner van de twaalfvingerige darm, worden symptomen gemanifesteerd door pijn in de epigastrische regio; misselijkheid, braken en winderigheid (duidt op darmobstructie); en, indien van aanzienlijke grootte, gastro-intestinale bloedingen. In gevallen van myoepitheliaal hamartoom van het ileum hebben patiënten klagen over buikpijn, hebben ze een verminderd lichaamsgewicht en ontwikkelen chronische anemie. [9], [10] ]
Lees ook - rectale poliepen
Retrorectaal hamartoma is een cystisch hamartoma of multichamberde cyste van de retrorectale ruimte (het losse bindweefsel tussen het rectum en zijn eigen fascia) dat meestal voorkomt bij vrouwen van middelbare leeftijd. Het heeft het uiterlijk van een cyste die uit de achterste wand van het rectum uitpuilt, die is bekleed met epitheel en chaotisch gerangschikte gladde spiervezels bevat. Dit hamartoma presenteert met lagere buikpijn en terugkerende constipatie. [11], [12] ]
Hamartomen van de lever en milt
Meerdere galhamartoma van de lever is een hamartoma van de interdollische intrahepatische galwegen geassocieerd met misvormingen van hun ontwikkeling tijdens de embryonale periode. Dit hamartoma (enkele of meerdere) bestaat uit lukraak verwijde clusters van galwegen en fibrocollagene stroma. [13]
Biliaire hamartomen zijn asymptomatisch en worden meestal overigens ontdekt (tijdens radiologisch onderzoek of laparotomie). [14]
Een zeldzaam en vaak overigens gedetecteerd primair neoplasma van goedaardige karakter is een hamartoom van de milt, dat bestaat uit elementen van de rode pulp van de milt - in de vorm van een goed gedefinieerde homogene massa van vaste consistentie. Deze misvorming kan single of meerdere zijn; Bij het persen van het milt parenchym kan er een gevoel van ongemak en pijn zijn in het linker subcostale gebied. [15], [16]
Nierhamartomen
Het meest voorkomende hamartoma van de nier wordt gediagnosticeerd als angiomyolipoom van de nier, omdat deze goedaardige tumor bestaat uit volwassen vetweefsel met ingebedde gladde spiervezels en bloedvaten. Het vormt zich in tubereuze sclerose in 40-80% van de gevallen. Het vergroten van de grootte van hamartoma (meer dan 4-5 cm) leidt tot pijn en het verschijnen van bloed in de urine. [17], [18]
Hamartoma van de borst
De WHO-geaccepteerde diagnostische definities van borsthamartoma zijn termen zoals adenolipoma, chondrolipoma en myoïde hamartoma. Hoewel vaak fibroadenolipoom genoemd door mammologen, omdat de tumorvorming cellen bevat van vezelachtig, klier- en vetweefsel ingesloten in een dunne bindweefselcapsule met verschillende contouren. Focale verkalkingen kunnen worden waargenomen bij visualisatie. In dit geval zijn klinische manifestaties afwezig. [19], [20] ]
Lees ook - borsttumoren
Hamartomen van de hersenen
Een derde van de patiënten met tubereuze sclerose heeft een hersenhamartoom in de vorm van intracraniële corticale uitgroei of knobbeltjes in verschillende lobben - aan de grens van grijze en witte materie - of subependymale knobbeltjes langs de wanden van de hersenvenrices. Astrocytisch hamartoma, een subpendymaal gigantische celastrocytoom met corticale verstoring, dysmorfe neuronen en grote gliale cellen van het hersenparenchym (astrocyten), kunnen zich ook vormen. Symptomen van cerebrale hamartomen zijn aanvallen van aanvallen en mentale retardatie bij kinderen. [21], [22]
Een zeldzame misvorming die optreedt tijdens embryogenese en aanwezig is bij de geboorte is een hypothalamisch hamartoom, dat een massa heterotopische neuronen en gliacellen is. Naarmate de hersenen van het kind groeien, groeit de tumor maar verspreidt zich niet naar andere cerebrale gebieden. [23], [24]
Als hypertrofische weefsels worden gevormd in het voorste deel van de hypothalamus (tuber cinereum), waar de hypofyse zich eraan hecht, manifesteert misvorming de symptomen van centrale voortijdige seksuele ontwikkeling (vóór 8-9 jaar oud): uiterlijk van acne-straal, vroege ontwikkeling van milkaderen en vroege menarche in meisjes; Vroege schaamhaar en stemmutatie bij jongens.
Wanneer Hamartomas zich in het achterste deel van de hypothalamus vormt, kunnen er afwijkingen zijn in de elektrische activiteit van de hersenen, die in vroege kinderschoenen wordt gemanifesteerd door aanvallen, en in een later stadium (leeftijd 4 tot 7 jaar) door epilepsie met focale epileptische aanvallen met plotselinge lawaai en mate-lawage en cognitieve problemen.
Een hypofyse hamartoma is een sporadisch voorkomende goedaardig hypofyse adenoma.
Volwassenen van middelbare leeftijd met het Cowden-syndroom kunnen een zeldzame tumorachtige massa hebben, een hamartoom van het cerebellum, gediagnosticeerd als dysplastische cerebellaire gangliocytoom of Lhermitte-Duclos ziekte. Symptomen kunnen afwezig of manifest zijn als hoofdpijn, duizeligheid, verminderde coördinatie van bewegingen en verlamming van individuele schedelzenuwen.
Lymfeknoophamartoma
Wanneer de cellen van gladde spier- en vetweefsel, evenals bloedvaten en collageenstroma van inguinale, retroperitoneale, submandibulaire en cervicale lymfeklieren overgroei, een angiomyomateuze hamartoma van een lymfe knooppunt of nodulaire angiomyomateuze hamartoma wordt gevormd - met partiële of complete vervanging van zijn parchyma. [25], [26]
Een hamartoma van de huid
In aanwezigheid van tubereuze sclerose of neurofibromatose worden verschillende hamartomen van de huid waargenomen, meestal in de vorm van gehypopigmenteerde plekken; koffie- en melkvlekken; angiofibroma (op de wangen, kin, nasolabiale plooien); Shagreen-vlekken van verschillende lokalisaties (die bindweefsel nevi zijn); Vezelige plaques op het voorhoofd, hoofdhuid of nek.
Een zeldzame dermatologische manifestatie van tubereuze sclerose (vooral bij mannen) is folliculocystisch en collageen hamartoom, gekenmerkt door overvloedige collageenafzetting in de dermis, concentrische perifolliculaire fibrose en keratine-gevulde trechtervormige subcutane cysten die worden gezien bij histopathologisch onderzoek. [27]
Naar hamartomen bestaande uit melanocyten (cellen die de pigment melanine produceren), verwijzen de meeste experts ook naar verschillende melanocytische neoplasmata, met name congenitale melanocytaire nevi, die een abnormaliteit van embryogenese vertegenwoordigt.
In termen van etiologie zijn hamartomen bestaande uit vasculair weefsel ook hemangiomen van de huid.
Patiënten met het Peutz-Jeghers-Thuren-syndroom hebben hamartoom in de vorm van fragmentarische pigmentatie van de huid en slijmvliezen - lentiginose periorificialis
Gevallen van lineair papulair ectodermaal-mesodermaal hamartoma (hamartoma moniliformis) vertonen een lineair vleeskleurige papieruitslag op de kop, nek en bovenste borst.
En een sebocytisch hamartoma is een hamartoma van de talgklieren, lees meer in de publicatie - sebacous nevus.
Hamartoma van het oog
Gepigmenteerde hamartomateuze laesies van de iris in neurofibromatosis type 1 en Watson-syndroom - in de vorm van nodulaire clusters van dendritische melanocyten - worden gedefinieerd als iris hamartomen of Lisch-knobbeltjes. Ze zijn transparant (meestal geen invloed op het gezichtsvermogen) afgeronde koepelvormige geelbruine papels die uitsteken boven het oppervlak van de iris.
En patiënten met juveniele angiofibroma van de nasopharynx en familiale adenomateuze polyposis ontwikkelen vaak gecombineerd hamartoma van het netvlies- en retinale pigmente-epitheel-in de vorm van een zwarte vlek op het centrale (maculair) deel van het netvlies. [28]
Een hamartoma van de neus
Nasale hamartoma wordt gedefinieerd door specialisten als nasaal chondromenchym hamartoma of nasale chondroma, vanwege goedaardige proliferatie van ademhalingsepitheel, submucosale klieren en chondro-bone mesenchym. De klinische manifestaties zijn afhankelijk van de grootte en lokalisatie van de laesie en omvatten: nasale congestie, moeilijkheid in nasale ademhaling en borstvoeding bij zuigelingen, heldere waterige neusafvoer en nasale bloedingen. Een hamartoma kan met het kind groeien en zich in de oogbanen verspreiden, wat resulteert in voorwaartse of achterwaartse verplaatsing van de oogbol, strabismus of oculomotorische verstoringen. [29]
Een hamartoma bij een kind
Alle bovengenoemde hamartomateuze laesies van verschillende organen en anatomische structuren zijn aanwezig bij kinderen met overeenkomstige syndromen.
Pasgeborenen aanwezig met mesenchymale hamartoom van de borstwand of kraakbeenhamartoom van de rib, die solide immobiele massa's zijn als gevolg van de focale overgroei van normale skeletelementen met kraakbeen-, vasculaire en mesenchymale elementen. Dit hamartoma kan ademhalingsfalen veroorzaken en de ontwikkeling van het respiratoire noodsyndroom. Mesenchymale hamartoma van de lever is de tweede meest voorkomende goedaardige levertumor bij kinderen. Deze tumorachtige vorming (vaker gelokaliseerd in de rechter lob van het orgaan) bestaat uit cellen van mesenchymale stroma, hepatocyten en epitheelcellen van galwegen voering. Het klinische beeld omvat voelbare massa in de buikholte, anorexia en gewichtsverlies, en in het geval van een significante grootte (tot 10 cm en meer) omvat de tumor extrahepatische galwegen en inferieure vena cava, die leidt tot geelzucht en oedeem van onderste extremiteiten.
Een hamartoma is een aangeboren mesoblastisch nefroma (dat voorkomt bij 1 op de 200.000 zuigelingen) dat kan leiden tot buik in de pasgeborene met een voelbare massa van dichte consistentie in het rechter bovenste kwadrant van de buik. Baby's kunnen ook een snelle ondiepe ademhaling presenteren.
Zeldzame aangeboren anomalieën omvatten vezelachtig hamartoom van de kindertijd, die voorkomt bij kinderen in de eerste twee jaar van het leven en presenteert als een pijnloze nodulaire massa in de onderhuidse tissues van de axilla, nek, schouder en onderarm, rug en borst, dij, voet en externe geslachtsdelen.
Eccrine angiomateus hamartoma bij een kind kan aanwezig zijn bij de geboorte of manifest in de vroege kinderjaren. Deze goedaardige tumor van hamartomateuze aard heeft meestal het uiterlijk van blauwachtige of bruinachtige knobbeltjes en/of plaques die het gevolg zijn van proliferatie van eccrine zweetklierweefsel en capillairen in de middelste en diepe lagen van de dermis. Dit hamartoma kan gelokaliseerde hyperhidrose en verhoogde haargroei veroorzaken.
Complicaties en gevolgen
Over het algemeen wordt het ermee eens dat Hamartoma's zelden terugkeren of transformeren in kwaadaardige tumoren. Ze vertonen vaak weinig of geen symptomen en verdwijnen soms zelfs in de loop van de tijd. Maar in meer ernstige gevallen en afhankelijk van de vormingsplaats, kunnen deze misvormingen ernstige complicaties en gevolgen hebben.
Allereerst kan een hamartoma groeien tot zo'n grootte dat het op omliggende weefsels en organen begint te drukken, waardoor hun functies worden verstoord.
Cardiale hamartoom bij kinderen kan leiden tot aanhoudende hartritmeafwijkingen, klepdefecten en verminderde intracardiale bloedstroom met daaropvolgend congestief hartfalen.
Complicaties van hamartomateuze poliepen van het GI-kanaal zijn maagdarmbloeding, obstructie en intestinale intussusceptie (met mogelijke fatale uitkomst). En een groot nierhamartoma kan de scheur van de nier uitlokken.
Een hamartoma in de hersenen kan obstructief hydrocephalus syndroom veroorzaken.
In hypothalamische en hypofysehamartomen kan de productie van somatotrope hormoon (groeihormoon) worden aangetast, wat leidt tot de ontwikkeling van hypofysaal nanisme (hypopituïtarisme) bij kinderen. Hypothalamische hamartomen bij kinderen kunnen ook leiden tot drugsresistente epilepsie.
Complicaties van retinale pigmentepitheelhamartoom zijn beladen met retinale en/of optische zenuwdisfunctie, maculair oedeem, neovascularisatie van het choroïde en retinale detachement.
Diagnostics Hamartomen
Een belangrijk onderdeel van de diagnose van hamartomen en gerelateerde syndromen is de verzameling van anamnesis, inclusief familiegeschiedenis.
Laboratoriumtests omvatten bloedtesten: algemeen klinisch; Serum-elektrolyten; lymfocytenprofiel; calcium-, kalium-, fosfaat- en ureumniveaus; en leverfunctietests. Indien mogelijk wordt een fijn-naald aspiratie-lekbiopsie van de massa uitgevoerd, omdat histologisch onderzoek cruciaal is bij de diagnose en keuze van behandelingstactieken.
Instrumentele diagnostiek biedt visualisatie van hamartomateuze tumorachtige vorming en identificatie van de exacte lokalisatie ervan, waarvoor röntgenfoto's, angiografie, elektro-encefalografie (EEG), echografie (sonografie), CT (computertomografie), PET (positronemissietomografie), MRI (magnetische resonantie-beeldvorming) worden gebruikt.
Differentiële diagnose
Bij elke abnormale massa's is differentiële diagnose erg belangrijk. Aldus worden tuberculoma en hamartoma gedifferentieerd; Longhamartoom en primaire longkanker, bronchogene carcinoïde, metastatische ziekte. Hersenhamartoma moet worden onderscheiden van craniofaryngioom en hypothalamisch-chiasmatisch glioom. En de differentiële diagnose van hamartoom als aangeboren mesoblastische nefroma omvat Wilms-tumor (kwaadaardig nefroblastoom), helder celsarcoom van de nier en ossifying niertumor bij zuigelingen.
Met wie kun je contact opnemen?
Behandeling Hamartomen
Als het hamartoom asymptomatisch is en per ongeluk wordt ontdekt, is geen behandeling vereist, maar het is noodzakelijk om het "gedrag" en de toestand van de patiënt te controleren. In andere gevallen is therapie gericht op het verminderen van de intensiteit van de symptomen en het voorkomen van complicaties. Bij bijvoorbeeld hypothalamisch hamartoom met symptomen van voortijdige puberteit worden bepaalde medicijnen die de afgifte van bepaalde hormonen remmen voorgeschreven. Cardiale medicijnen worden gebruikt om symptomen van hartfalen te behandelen bij patiënten met harthamartomen.
Chirurgische verwijdering van hamartomen is geïndiceerd om de diagnose te bevestigen en in gevallen van medisch niet-correcteerbare intense symptomen.
Longhamartomen kunnen bijvoorbeeld worden geresecteerd door wigresectie en, in ernstige gevallen, door verwijdering van een lob van de long (lobectomie). Een borsthamartoma kan ook worden uitgesneden, en als het groot is, kan een gedeeltelijke of volledige borstamputatie vereist zijn.
Stereotactische radiofrequente thermoablatie of laserablatie kan worden gebruikt om hamartomateuze poliepen te verwijderen. Radioschirurgie met sterk gerichte gammastralen - gamma-mes voor hypothalamische hamartomen of astrocytische hamartomen - wordt ook gebruikt.
Het voorkomen
De enige methode om de ontwikkeling van hamartoma's te voorkomen, kan worden beschouwd genetische screening van de toekomstige ouders van het kind.
Prognose
De algehele prognose van deze aangeboren anomalie hangt af van de lokalisatie en grootte van het neoplasma, evenals van comorbiditeiten en de algemene gezondheid van de patiënt.