Medisch expert van het artikel

Nieuwe publicaties
Keratodermie: oorzaken, symptomen, diagnose, behandeling
Laatst beoordeeld: 07.07.2025

Alle iLive-inhoud wordt medisch beoordeeld of gecontroleerd op feiten om zo veel mogelijk feitelijke nauwkeurigheid te waarborgen.
We hebben strikte richtlijnen voor sourcing en koppelen alleen aan gerenommeerde mediasites, academische onderzoeksinstellingen en, waar mogelijk, medisch getoetste onderzoeken. Merk op dat de nummers tussen haakjes ([1], [2], etc.) klikbare links naar deze studies zijn.
Als u van mening bent dat onze inhoud onjuist, verouderd of anderszins twijfelachtig is, selecteert u deze en drukt u op Ctrl + Enter.
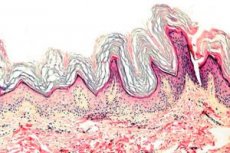
Keratodermie is een groep dermatosen die gekenmerkt worden door een verstoring van het keratinisatieproces: overmatige hoornvorming, voornamelijk op de handpalmen en voetzolen.
De oorzaken en pathogenese van de ziekte zijn nog niet volledig opgehelderd. Onderzoek heeft aangetoond dat keratodermieën worden veroorzaakt door mutaties in de genen die coderen voor keratine 6, 9 en 16. Vitamine A-tekort, hormonale disfuncties, voornamelijk van de geslachtsklieren, en bacteriële en virale infecties spelen een grote rol in de pathogenese. Ze behoren tot de symptomen van erfelijke aandoeningen en tumoren van inwendige organen (parapsoriatische keratodermieën).
Symptomen. Er wordt onderscheid gemaakt tussen diffuse (Unna-Tost keratodermie, Meleda keratodermie, Papillon-Lefevre keratodermie, mutilerende keratodermie en syndromen waarbij diffuse keratodermie een van de hoofdsymptomen is) en focale (dissemineerde gevlekte keratodermie van Fischer-Buschke, acrokeratoelastoïdose van Kosti, beperkte keratodermie van Bruhauer-Franzesthesti, lineaire keratodermie van Fuchs, etc.) keratodermie.
Winy-Tost keratodermie (synoniemen: congenitale ichthyosis van de handpalmen en voetzolen, Winy-Tost syndroom) wordt autosomaal dominant overgedragen. Er is sprake van een diffuse overmatige keratinisatie van de huid van de handpalmen en voetzolen (soms alleen de voetzolen), die zich ontwikkelt in de eerste twee levensjaren. Het huidpathologische proces begint met een lichte verdikking van de huid van de handpalmen en voetzolen in de vorm van een strook erytheem met een fuchsiale kleur op de grens met gezonde huid. Na verloop van tijd verschijnen gladde, gelige hoornlagen op het oppervlak. De laesie verspreidt zich zelden naar de rug van de polsen of vingers. Bij sommige patiënten kunnen zich oppervlakkige of diepe kloven vormen en wordt lokale hyperhidrose opgemerkt. Bij de door de auteur geobserveerde patiënt, de oom van moederskant, broer en zoon, leden Winy-Tost keratodermie.
Er worden gevallen beschreven van schade aan nagels (verdikking), tanden en haar bij Winy-Tost keratoderma in combinatie met verschillende skeletafwijkingen en pathologieën van inwendige organen, zenuw- en endocriene systemen.
Histopathologie. Histologisch onderzoek toont duidelijke hyperkeratose, granulose, acanthose en kleine ontstekingsinfiltraten in de bovenste dermis. Differentiële diagnose. De ziekte moet worden onderscheiden van andere vormen van keratodermie.
Meleda keratodermie (synoniemen: ziekte van Meleda, congenitaal progressief acrokeratoom, Siemens palmoplantaire transgradiënte keratose, Kogoy's erfelijke palmoplantaire progressieve keratose) erft autosomaal recessief over. Deze vorm van keratodermie wordt gekenmerkt door dikke, geelbruine hoornlagen met diepe barstjes. Een violetpaarse rand van enkele millimeters breed is zichtbaar langs de randen van de laesie. Het proces breidt zich meestal uit naar de ruggen van handen en voeten, onderarmen en schenen. De meeste patiënten ervaren lokale hyperhidrose. Hierbij worden de handpalmen en voetzolen licht vochtig en bedekt met zwarte puntjes (zweetklierkanalen).
De ziekte kan zich ontwikkelen tussen de leeftijd van 15 en 20 jaar. Nagels worden dikker en misvormd.
Histopathologie. Histologisch onderzoek toont hyperkeratose, soms acanthose en een chronisch ontstekingsinfiltraat in de papillaire dermis.
Differentiële diagnose. Melela keratodermie moet worden onderscheiden van Unna-Tost keratodermie.
Keratoderma Papillon-Lefevre (synoniem: palmoplantaire hyperkeratose met parodontitis) erft autosomaal recessief over.
De ziekte manifesteert zich in het tweede tot derde levensjaar. Het klinische beeld van de ziekte is vergelijkbaar met dat van de ziekte van Melela. Daarnaast zijn veranderingen in het gebit kenmerkend (afwijkingen bij de doorbraak van melk- en blijvende tanden met cariësvorming, gingivitis, snel progressieve parodontitis met vroegtijdig tandverlies).
Histopathologie. Histologisch onderzoek toont verdikking van alle lagen van de opperhuid, met name de hoornlaag, en onbeduidende celclusters van lymfocyten en histiocyten in de lederhuid.
Differentiële diagnose. De ziekte moet worden onderscheiden van andere keratodermieën. Een belangrijk onderscheidend kenmerk is de karakteristieke dentale pathologie, die niet voorkomt bij andere vormen van erfelijke diffuse keratodermieën.
Keratoderma mutilans (synoniemen: syndroom van Fonwinkel, erfelijk mutilerend keratoom) is een vorm van diffuse keratodermie die autosomaal dominant overerft. Het ontwikkelt zich in het tweede levensjaar en wordt gekenmerkt door diffuse hoornachtige afzettingen op de huid van handpalmen en voetzolen met hyperhidrose. Na verloop van tijd vormen zich koordachtige groeven op de vingers, wat leidt tot contracturen en spontane amputatie van de vingers. Folliculaire keratose manifesteert zich op de handruggen, evenals in het gebied van de elleboog- en kniegewrichten. De nagelplaten zijn veranderd (vaak als horlogeglazen). Er zijn gevallen beschreven van hypogonadisme, robijnrode alopecia, gehoorverlies en pachyonychia.
Histopathologie. Histologisch onderzoek toont ernstige hyperkeratose, granulose, acanthose en kleine ontstekingsinfiltraten in de dermis, bestaande uit lymfocyten en histiocyten.
Differentiële diagnose. Bij het onderscheiden van mutilerende keratodermie van andere vormen van diffuse keratodermie moet allereerst rekening worden gehouden met het mutilerende effect, dat niet kenmerkend is voor andere vormen. Bij het uitvoeren van differentiële diagnostiek van alle vormen van diffuse keratodermie is het belangrijk om te onthouden dat dit een van de hoofdsymptomen kan zijn van een aantal erfelijke syndromen.
Behandeling. Neotigazon is geïndiceerd voor de algemene behandeling van keratodermie. De dosering van het geneesmiddel is afhankelijk van de ernst van het proces en bedraagt 0,3-1 mg/kg lichaamsgewicht. Bij afwezigheid van neotigazon wordt vitamine A aanbevolen in een dosis van 100 tot 300.000 mg per dag gedurende een lange periode. Externe therapie bestaat uit het gebruik van zalven met aromatische retinoïden, keratolytische en steroïde middelen.
 [
[ Wat zit je dwars?
Wat moeten we onderzoeken?
Hoe te onderzoeken?