Medisch expert van het artikel

Nieuwe publicaties
Amaurotische idiotie
Last reviewed: 04.07.2025

Alle iLive-inhoud wordt medisch beoordeeld of gecontroleerd op feiten om zo veel mogelijk feitelijke nauwkeurigheid te waarborgen.
We hebben strikte richtlijnen voor sourcing en koppelen alleen aan gerenommeerde mediasites, academische onderzoeksinstellingen en, waar mogelijk, medisch getoetste onderzoeken. Merk op dat de nummers tussen haakjes ([1], [2], etc.) klikbare links naar deze studies zijn.
Als u van mening bent dat onze inhoud onjuist, verouderd of anderszins twijfelachtig is, selecteert u deze en drukt u op Ctrl + Enter.

Amaurotische idiotie is een zeldzame, progressieve ziekte. Deze wordt gekenmerkt door een geleidelijke afname van het gezichtsvermogen tot volledige blindheid en een afname van de intelligentie, totdat de idiotie zich manifesteert. Als gevolg hiervan ontwikkelt de patiënt ernstige marasmus met een fatale afloop. De ziekte werd meer dan 130 jaar geleden voor het eerst beschreven door oogarts Dr. Tau. Tau constateerde een bijzondere transformatie van de fundus. Er zijn al meer dan 500 gevallen van de ziekte beschreven.
 [ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ]
[ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ]
Epidemiologie
Oorzaken amaurotische idiotie
Ondanks de vele gegevens die over de ziekte zijn verzameld, blijven wetenschappers momenteel zoeken naar antwoorden op veel vragen over de oorzaken, pathogenese en zelfs verschijnselen van amaurotische idiotie.
Er zijn aanwijzingen dat de ziekte erfelijk is. De overerving is autosomaal recessief. Meestal worden de kleine hersenen en de achterhoofdskwabben van de hersenhelften aangetast, met ernstige gevolgen en complicaties voor het hele lichaam: atrofie van de oogzenuwen, zenuwvezels kunnen hun membranen verliezen en verbindingen tussen zenuwcellen kunnen verbrokkelen.
De meeste deskundigen erkennen dat de klinische symptomen van de ziekte zeer uiteenlopend kunnen zijn en dat deze samenhangen met de leeftijd waarop amaurotische idiotie zich bij de patiënt begon te ontwikkelen.
Tijdens het onderzoek naar de oorzaken van de ziekte werd een bepaald patroon opgemerkt: de ziekte treft vaak kinderen uit hetzelfde gezin, vandaar de naam "familiale amaurotische idiotie". Volgens de studies, waarvan de resultaten werden gepubliceerd toen de studie naar de ziekte nog maar net begon, werden van de 64 gevallen van amaurotische idiotie er 37 aangetroffen in 13 gezinnen (elk gezin had 2-5 zieke kinderen). Het is opmerkelijk dat in dergelijke gezinnen de zieken volkomen gezonde broers en zussen hadden. Tegenwoordig wordt aangenomen dat recessieve overerving een grote rol speelt bij het ontstaan van de ziekte. Zo is het mogelijk om de frequentie van het voorkomen van gevallen van de ziekte in dezelfde gezinnen te verklaren. Bij het analyseren van de erfelijkheidsfactor als oorzaak van amaurotische idiotie mag men zich niet beperken tot de aanwezigheid van klinisch tot uiting komende tekenen in families van patiënten (zowel in de opgaande als in de laterale lijnen), maar moet men ook rekening houden met rudimentaire tekenen, bijvoorbeeld karakteristieke afwijkingen in de werking van het visuele apparaat (familiaire choroïditis, pigmentaire retinale dystrofie, enz.).
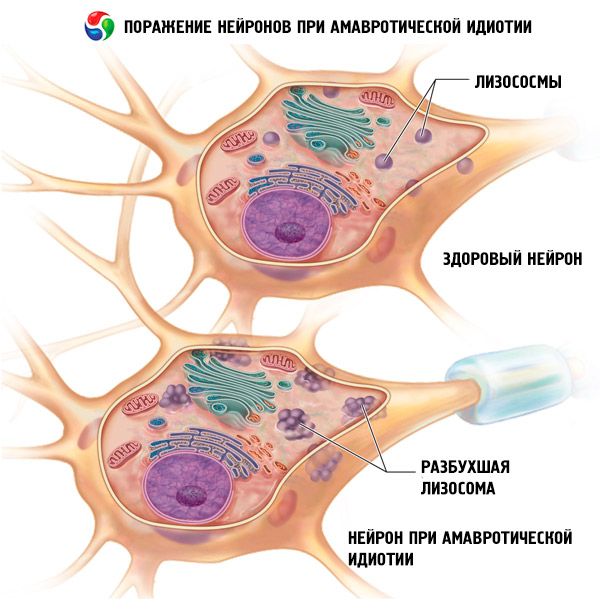
Symptomen amaurotische idiotie
De eerste tekenen van de aangeboren vorm openbaren zich in de eerste dagen of weken van het leven. De baby wordt geboren met hydrocefalie of microcefalie, lijdt aan epileptische aanvallen, verlamming en ademhalingsmoeilijkheden. Het kind overlijdt na enkele maanden.
Stages
De infantiele vorm ontwikkelt zich tussen de 4 en 6 maanden. Deze vorm van amaurotische idiopathieën wordt gekenmerkt door een familiaire aard. Het gezichtsvermogen neemt snel af: de baby kan zijn blik niet fixeren en kan geen objecten waarnemen. Op de fundus verschijnt de zogenaamde "kersenpit" - een roodachtige vlek in de macularegio, omgeven door een grijswitte rand. Vervolgens atrofiëren de oogzenuwen en verliest het kind volledig het gezichtsvermogen. Oriëntatie, beschermende reflexen en het bewegingsvermogen gaan geleidelijk verloren. Patiënten reageren sterk op geluidsstimuli - ze deinzen terug voor een geluid dat voor een gezond persoon zacht is, en convulsies kunnen worden waargenomen als gevolg van een verhoogde spierspanning. In het laatste stadium van de ziekte ontwikkelen zich algemene atrofie, uitputting van het lichaam en een verhoogde spanning van alle strekspieren. De prognose van de ziekte is eveneens teleurstellend: de patiënt overlijdt anderhalf tot twee jaar na het begin van de ziekte.
De late kindertijdvorm begint rond de leeftijd van 3-4 jaar. De progressieve ziekte wisselt af met stadia van remissie. Het geleidelijke verlies van intelligentie gaat gepaard met epileptische aanvallen, coördinatiestoornissen en extrapiramidale stoornissen. Deze vorm wordt ook gekenmerkt door atrofie van de oogzenuw. De dood treedt 6-8 jaar na het begin van de amaurotische idiotie in.
De juveniele vorm begint zich te manifesteren tussen de 6 en 10 jaar. De amaurotische idiotie van Spielmeyer ontwikkelt zich minder snel. Veranderingen in de fundus vallen samen met manifestaties van pigmentaire retinale dystrofie. Het gezichtsvermogen van de patiënt neemt geleidelijk af, evenals de intelligentie. Verminderde motorische functies kunnen zich op verschillende manieren en onregelmatig manifesteren: weinig uitgesproken verlammingen van armen en benen, extrapiramidale en bulbaire stoornissen komen voor. De ziekte leidt 10 tot 25 jaar na het ontstaan van de eerste tekenen tot de dood.
De late vorm komt zeer zelden voor en ontwikkelt zich extreem langzaam. De mentale toestand van de patiënt verandert (zoals bij een organisch mentaal syndroom), er is sprake van atrofie van de oogzenuwen en pigmentdystrofie van het netvlies. Het laatste stadium wordt gekenmerkt door verlamming en een epileptiform syndroom. De patiënt overlijdt 10-15 jaar na het begin van de ziekte.
[ 38 ], [ 39 ], [ 40 ], [ 41 ], [ 42 ], [ 43 ], [ 44 ], [ 45 ]
Vormen
Er zijn vier soorten amaurotische idiotie:
- Tay-Sachs (treedt op jonge leeftijd op);
- Jansky-Bilynovsky (verschijnt bij kinderen op latere leeftijd);
- Spielmeyer-Vogt-syndroom (komt voor bij adolescenten);
- Kufsa (late vorm).
Sommige wetenschappers maken ook een apart onderscheid tussen het aangeboren Norman-Wood-type.
Elk type ziekte kent zijn eigen klinische verschijnselen, maar ze hebben allemaal gemeenschappelijke oorzaken, een gemeenschappelijk klinisch beeld, een gemeenschappelijke anatomische basis en een gemeenschappelijke pathogenese.
[ 46 ], [ 47 ], [ 48 ], [ 49 ], [ 50 ], [ 51 ], [ 52 ], [ 53 ], [ 54 ]
Diagnostics amaurotische idiotie
Amaurotische idiotie wordt veroorzaakt door een stoornis in de lipidenstofwisseling, waarbij een tussenproduct van de lipidenstofwisseling, sfingomyeline, zich afzet in verschillende lichaamscellen. De locatie en samenstelling van de afzettingen bepalen de ontwikkeling van een specifiek klinisch beeld van de ziekte.
[ 55 ], [ 56 ], [ 57 ], [ 58 ], [ 59 ], [ 60 ], [ 61 ], [ 62 ], [ 63 ]
Hoe te onderzoeken?
Differentiële diagnose
De differentiële diagnose van amaurotische idiotie is gebaseerd op een specifiek klinisch beeld en karakteristieke pathologieën van het fundus.
De vroege vorm vertoont symptomen die lijken op de ziekte van Landing, een vorm van mucopolysacharidose. De ziekte van Landing ontwikkelt zich vanaf de eerste maanden na de geboorte en leidt na 2-3 jaar tot de dood. In 1/5 van de gevallen verschijnt een "kersenpit" op de fundus, degeneratieve veranderingen in het netvlies en een vervormde waarneming van geluiden (hypercussie) zijn er vrijwel niet kenmerkend voor, maar gelijktijdige vergroting van de milt en lever, psychische stoornissen en bewegingsstoornissen worden wel opgemerkt.
De juveniele vorm overlapt soms de manifestaties van het Lawrence-Moon-Biedl-syndroom. Om deze ziekten te differentiëren, is het noodzakelijk om aandacht te besteden aan hun andere manifestaties. Het Lawrence-Moon-Biedl-syndroom wordt gekenmerkt door snelle gewichtstoename, misvorming van de ledematen, gekenmerkt door de aanwezigheid van extra vingers of tenen, opvallende vegetatief-trofische stoornissen en de afwezigheid van motorische functiestoornissen.
De verscheidenheid aan symptomen van late amaurotische idiotie compliceert de diagnose tijdens het leven. De manifestaties ervan lijken op die van Friedreichs ataxie, multiple sclerose, de ziekte van Alzheimer, de ziekte van Pick, progressieve verlamming en zelfs schizofrenie.
Sommige auteurs benadrukken dat de diagnose van deze ziekte, vooral wanneer de klinische verschijnselen onduidelijk zijn, pas na overlijden met zekerheid kan worden gesteld, op basis van de analyse van histologische afwijkingen van het zenuwstelsel.
Met wie kun je contact opnemen?
Behandeling amaurotische idiotie
Er is geen rationele en effectieve behandeling. Tegenwoordig is de therapie voor amaurotische idiotie uitsluitend gericht op symptoomverlichting. Er worden kalmerende middelen, nootropica, anti-epileptica en algemene tonica gebruikt.
Om de bloedsomloop en de stofwisselingsprocessen in de hersenen te activeren, worden glycine, elkar, cerebrolysine, glutaminezuur en pantogam voorgeschreven.
Om het convulsief syndroom te verlichten, worden difenine of carmazepine voorgeschreven.
Een positief resultaat kan bereikt worden door gebruik te maken van weefselextracten, bloedtransfusie of plasma.
Het voorkomen
Het gebrek aan effectieve therapie voor amaurotische idiotie dwingt ons tot intensieve aandacht voor preventie. Er bestaan al methoden om heterozygote dragers van het pathologische gen te identificeren en methoden om amaurotische idiotie tijdens de zwangerschap te diagnosticeren. Prenatale diagnostiek van de ziekte bestaat uit het analyseren van de activiteit van hexosaminidase A in het vruchtwater. Indien een verminderde enzymactiviteit wordt vastgesteld, is het raadzaam de zwangerschap te beëindigen. Ouders van een ziek kind wordt geadviseerd geen kinderen meer te krijgen.
Использованная литература