Medisch expert van het artikel

Nieuwe publicaties
Polymyositis en dermatomyositis: oorzaken, symptomen, diagnose, behandeling
Laatst beoordeeld: 23.04.2024

Alle iLive-inhoud wordt medisch beoordeeld of gecontroleerd op feiten om zo veel mogelijk feitelijke nauwkeurigheid te waarborgen.
We hebben strikte richtlijnen voor sourcing en koppelen alleen aan gerenommeerde mediasites, academische onderzoeksinstellingen en, waar mogelijk, medisch getoetste onderzoeken. Merk op dat de nummers tussen haakjes ([1], [2], etc.) klikbare links naar deze studies zijn.
Als u van mening bent dat onze inhoud onjuist, verouderd of anderszins twijfelachtig is, selecteert u deze en drukt u op Ctrl + Enter.
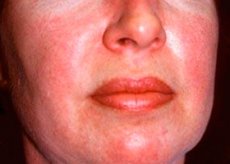
Poliomyositis en dermatomyositis zijn zeldzame systemische reumatische ziekten die worden gekenmerkt door inflammatoire en degeneratieve spierveranderingen (polymyositis) of spieren en huid (dermatomyositis). De meest specifieke huidmanifestatie is een heliotrope uitslag.
De spierlaesies zijn symmetrisch en omvatten zwakte, enige pijn en daaropvolgende atrofie van de proximale spieren van de bovenste extremiteitsriem. Complicaties kunnen interne orgaanschade en maligniteit omvatten. De diagnose is gebaseerd op de analyse van het klinische beeld en de evaluatie van spieraandoeningen door de concentraties van de relevante enzymen te bepalen, MRI, elektromyografie en biopsie van spierweefsel uit te voeren. De behandeling maakt gebruik van glucocorticoïden, soms in combinatie met immunosuppressiva of immunoglobulinen die intraveneus worden toegediend.
Vrouwen zijn tweemaal zo vaak ziek als mannen. De ziekte kan op elke leeftijd voorkomen, maar wordt vaker gedetecteerd in het interval van 40 tot 60 jaar; bij kinderen van 5 tot 15 jaar.
 [
[Wat veroorzaakt dermatomyositis en polymyositis?
De oorzaak van de ziekte is een auto-immuunreactie op spierweefsel bij mensen met genetische aanleg. De ziekte komt vaker voor in de aanwezigheid van een belaste familiegeschiedenis en dragers van sommige HLA-antigenen (DR3, DR52, DR56). Mogelijke opstartfactoren zijn virale myositis en maligne neoplasmata. Er zijn meldingen van de detectie in spiercellen van structuren vergelijkbaar met picornavirussen; Bovendien kunnen virussen bij dieren vergelijkbare ziekten veroorzaken. Maligniteiten associatie met dermatomyositis (veel minder dan bij polymyositis) suggereert dat tumorgroei ook een startmechanisme ziekten als gevolg van autoimmuniteit run op gedeelde tumorantigenen en spierweefsel zijn.
In de wanden van de bloedvaten van skeletspieren worden afzettingen van IgM, IgG en de derde complementcomponent gedetecteerd; dit geldt vooral voor kinderen met dermatomyositis. Patiënten met polymyositis kunnen ook andere auto-immuunprocessen ontwikkelen.
Pathofysiologie dermatomyositis en polymyositis
Pathologische veranderingen omvatten celbeschadiging en hun atrofie tegen een achtergrond van ontsteking van variërende ernst. De spieren van de bovenste en onderste ledematen, evenals het gezicht, zijn minder aangetast dan andere skeletspieren. De nederlaag van de viscerale musculatuur van de keelholte en de bovenste delen van de slokdarm, minder vaak van het hart, de maag of de darm, kan leiden tot verstoring van de functies van deze organen. Hoge concentraties myoglobine, veroorzaakt door rabdomyolyse, kunnen nierbeschadiging veroorzaken. Er kunnen ook ontstekingsveranderingen in de gewrichten en longen optreden, vooral bij patiënten met antisynthetische antilichamen.
Symptomen van dermatomyositis en polymyositis
Het begin van polymyositis kan acuut (vooral bij kinderen) of subacuut (meestal bij volwassenen) zijn. Een acute virale infectie gaat soms vooraf of is de startfactor van de manifestatie van de ziekte, waarvan de meest frequente manifestaties de zwakte zijn van de proximale spieren of huiduitslag. Pijngevoelens worden minder uitgedrukt dan zwakte. Misschien de ontwikkeling van polyartralgia, het fenomeen van Raynaud, dysfagie, longenongevallen, veel voorkomende symptomen (koorts, verlaging van de massa, zwakte). Het fenomeen van Reynaud wordt vaak gevonden bij patiënten die gelijktijdig bindweefselaandoeningen hebben.
Spierzwakte kan enkele weken of maanden duren. Voor de klinische manifestatie van spierzwakte moet echter minimaal 50% van de spiervezels worden aangetast (dus de aanwezigheid van spierzwakte wijst op progressie van de myositis). Patiënten kunnen moeilijkheden ondervinden om hun armen boven schouderhoogte op te tillen, de trap op te lopen, op te staan van de vergadering. Vanwege de uitgesproken zwakte van de bekkenspieren en de schoudergordel, kunnen patiënten worden vastgeklonken aan een rolstoel of bed. Met het verslaan van de flexor van de nek wordt het onmogelijk om het hoofd van het kussen te scheuren. Het verslaan van de spieren van de keelholte en de bovenste delen van de slokdarm leidt tot een schending van slikken en regurgitatie. De spieren van de onderste ledematen en het aangezicht worden meestal niet beïnvloed. De ontwikkeling van contracturen van ledematen is echter mogelijk.
Huiduitslag, genoteerd met dermatomyositis, heeft meestal een donkere kleur en erythemateuze karakter. Periorbitale zwelling van paarse kleur (heliotrope huiduitslag) is ook kenmerkend. Huiduitbarstingen kunnen enigszins boven het huidniveau uitstijgen en glad zijn of bedekt met schubben; localisatie van laesies - het voorhoofd, nek, schouders, borst, rug, onderarmen, onderste delen van de dijen wenkbrauwen, knie, de mediale malleolus, het achteroppervlak van de interfalangeale en metacarpofalangeale gewrichten, de laterale zijde (Gottrona symptoom). Mogelijke hyperemie van de basis of periferie van de nagels. Op de huid van het laterale oppervlak van de vingers is het mogelijk om desquamative dermatitis te ontwikkelen, vergezeld van scheuren. Primaire huidletsels vaak opgelost zonder gevolgen, maar kan leiden tot de ontwikkeling van secundaire veranderingen in de vorm van donkere pigmentatie, atrofie, littekens, of vitiligo. Mogelijk de vorming van onderhuidse calcificaties, vooral bij kinderen.
Ongeveer 30% van de patiënten ontwikkelt polyarthralgie of polyartritis, vaak gepaard gaand met zwelling en gewrichtseffusie. Niettemin is de ernst van de uitwendige gewrichten klein. Vaker komen ze voor wanneer patiënten antilichamen tegen Jo-1 of andere synthetasen hebben.
Het verslaan van de interne organen (met uitzondering van de farynx en de bovenste slokdarm) bij polymyositis komt minder vaak voor dan bij andere reumatische aandoeningen (in het bijzonder SLE en systemische sclerodermie). Zelden, vooral met een antisynthetisch syndroom, manifesteert de ziekte zich als een interstitiële pneumonitis (in de vorm van dyspnoe en hoest). Hartritmestoornissen en geleidingsstoornissen kunnen zich ontwikkelen, maar ze zijn meestal asymptomatisch. Manifestaties uit het maagdarmkanaal komen vaker voor bij kinderen die ook last hebben van vasculitis en kunnen braken omvatten met een mengsel van bloed, melena en perforatie van de darm.
Waar doet het pijn?
Classificatie van polymyositis
Er zijn 5 varianten van polymyositis.
- Primaire idiopathische polymyositis, die op elke leeftijd kan voorkomen. Hiermee is er geen huidletsel.
- Primaire idiopathische dermatomyositis is vergelijkbaar met de primaire idiopathische polymyositis, maar daarmee is er een laesie van de huid.
- Polymyositis en dermatomyositis geassocieerd met maligne neoplasmata kunnen optreden bij patiënten van elke leeftijd; het vaakst waargenomen bij oudere patiënten, evenals bij patiënten met andere bindweefselaandoeningen. De ontwikkeling van maligne neoplasmata kan zowel binnen 2 jaar vóór als binnen 2 jaar na het debuut van myositis worden waargenomen.
- Kinder polymyositis of dermatomyositis wordt geassocieerd met systemische vasculitis.
- Polymyositis en dermatomyositis kunnen ook voorkomen bij patiënten die lijden aan andere bindweefselaandoeningen, meestal met progressieve systemische sclerose, gemengde bindweefselziekte en SLE.
Opname van de spier van de romp in de groep van polymyositis van myositis is onjuist, omdat de laatste een afzonderlijke ziekte is die wordt gekenmerkt door klinische manifestaties die vergelijkbaar zijn met die van chronische idiopathische polymyositis. Het ontwikkelt zich echter op oudere leeftijd, beïnvloedt vaak de spieren van de distale delen van het lichaam (bijvoorbeeld de bovenste en onderste ledematen), heeft een langere duur, reageert slecht op de behandeling en wordt gekenmerkt door een typisch histologisch beeld.
Diagnose van dermatomyositis en polymyositis
Poliomyositis moet vermoed worden bij patiënten met klachten van zwakte van de proximale spieren, vergezeld van hun pijn of zonder. Testen op dermatomyositis vereist bij patiënten klaagden over een uitslag die lijkt heliotrope of symptoom Gottrona, alsmede bij patiënten met symptomen van polymyositis, gecombineerd met huidlaesies passende dermatomyositis. De klinische manifestaties van polymyositis en dermatomyositis kunnen lijken op die van systemische sclerose of, minder vaak - SLE of vasculitis. De betrouwbaarheid van de diagnose wordt verhoogd door een zo groot mogelijk aantal van deze vijf criteria te matchen:
- zwakte van proximale spieren;
- kenmerkende huiduitslag;
- verhoogde activiteit van spierweefsel enzymen (creatine kinase of, bij afwezigheid van een toename van de activiteit, aminotransferasen of aldolase);
- karakteristieke veranderingen in myografie of MRI;
- kenmerkende histologische veranderingen in de biopsie van spierweefsel (absoluut criterium).
Een spierbiopsie kan sommige klinisch vergelijkbare aandoeningen uitsluiten, zoals spiertorso-spier en rhabdomyolyse veroorzaakt door een virale infectie. De veranderingen die worden onthuld door histologisch onderzoek kunnen verschillen, maar typisch zijn chronische ontstekingen, foci van degeneratie en regeneratie van spieren. Vóór het begin van potentieel toxische behandeling moet een nauwkeurige diagnose worden gesteld (meestal door middel van histologische verificatie). Met MRI is het mogelijk om brandpunten van oedeem en ontsteking in de spieren te identificeren, gevolgd door een gerichte biopsie.
Laboratoriumstudies laten versterken of juist te elimineren verdenking op de aanwezigheid van ziekten, en zijn ook bruikbaar bij het evalueren van de ernst ervan, mogelijke combinatie met andere diagnostische en pathologie soortgelijke complicaties. Ondanks het feit dat bij sommige patiënten antinucleaire antilichamen worden gedetecteerd, is dit fenomeen meer typerend voor andere bindweefselaandoeningen. Ongeveer 60% van de patiënten heeft antilichamen tegen het nucleaire antigeen (PM-1) of hele cellen van de thymus en Jo-1. De rol van auto-antilichaam de pathogenese van de ziekte blijft onduidelijk, hoewel het bekend dat antilichamen tegen Jo-1 specifieke merker antisintetaznogo syndroom omvattende fibroserende alveolitis, longfibrose, artritis, Raynaud-fenomeen.
Periodieke beoordeling van creatinekinase-activiteit is nuttig voor het bewaken van de behandeling. Niettemin, met ernstige spieratrofie, kan de activiteit van het enzym normaal zijn, ondanks de aanwezigheid van chronische actieve myositis. MRI-gegevens, spierbiopten of hoge waarden van creatinekinase-activiteit helpen vaak om het terugkeren van polymyositis en myopathie veroorzaakt door glucocorticoïden te differentiëren.
Omdat veel patiënten gediagnosticeerd kanker, raden sommige auteurs screening van alle volwassenen met dermatomyositis, en personen die lijden aan polymyositis, op de leeftijd van 60 als volgt: lichamelijk onderzoek, vkpyuchayuschy borstonderzoek, onderzoek van het bekken en de inspectie van de endeldarm ( inclusief de studie van uitwerpselen voor latent bloed); klinische bloedtest; biochemische bloedonderzoeken; mammografie; bepaling van het kanker-embryonale antigeen; algemene analyse van urine; thoraxfoto. De behoefte aan dergelijke screening voor jongere patiënten die geen klinische tekenen van kwaadaardige tumoren hebben, werd door sommige auteurs in twijfel getrokken.
Wat moeten we onderzoeken?
Hoe te onderzoeken?
Behandeling van dermatomyositis en polymyositis
Alvorens een ontsteking te stoppen, is het noodzakelijk om fysieke activiteit te beperken. Glucocorticoïden zijn eerstelijnsgeneesmiddelen. In de acute fase van de ziekte hebben volwassen patiënten prednisolon (binnen) nodig in een dosis van 40 tot 60 mg per dag. Regelmatige detectie van creatinekinase-activiteit is een vroege indicator van de werkzaamheid: bij de meeste patiënten vindt de afname of normalisatie plaats binnen 6 tot 12 weken na de toename van de spierkracht. Na de normalisatie van de enzymactiviteit neemt de dosis prednisolon af: eerst ongeveer 2,5 mg per dag gedurende de week, daarna sneller; wanneer de activiteit van de spier-enzymen toeneemt, neemt de dosis van het hormoon opnieuw toe. Herstelde patiënten kunnen het doen zonder glucocorticoïden, maar vaker hebben volwassen patiënten langdurige glucocorticoïde therapie nodig (10-15 mg prednisolon per dag). De aanvangsdosis prednisolon voor kinderen is eenmaal daags 30-60 mg / m 2. Als er een remissie is> 1 jaar, kunnen kinderen stoppen met de behandeling met glucocorticoïden.
In sommige gevallen, bij patiënten die hoge doses glucocorticoïden krijgen, treedt plotselinge toename van spierzwakte op, wat mogelijk te wijten is aan de ontwikkeling van glucocorticoïde myopathie.
Bij ontoereikende reactie op glucocorticoïde behandeling en ook in de ontwikkeling van glucocorticoïde myopathie of andere complicaties die dosisverlaging of intrekking prednisolon, gebruikt immunosuppressiva (methotrexaat, cyclofosfamide, azathioprine, cyclosporine). Sommige patiënten kunnen alleen methotrexaat krijgen (meestal in doses hoger dan die in de behandeling van RA) voor meer dan 5 jaar. Intraveneuze immunoglobulines effectief kan zijn bij patiënten die ongevoelig zijn voor medicamenteuze behandeling te zijn, maar het gebruik ervan verhoogt de kosten van de behandeling.
Myositis geassocieerd met primaire en gemetastaseerde tumoren, evenals myositis van de rompspieren, zijn meestal meer ongevoelig voor behandeling met glucocorticoïden. De ontwikkeling van remissie geassocieerd met kwaadaardige tumoren van myositis is mogelijk na verwijdering van de tumor.
Welke prognose heeft dermatomyositis en polymyositis?
Langdurige remissie (en zelfs klinisch herstel) gedurende 5 jaar worden opgemerkt bij meer dan de helft van de behandelde patiënten; bij kinderen is deze indicator hoger. Terugval kan zich echter op elk moment ontwikkelen. De totale vijfjaarsoverleving is 75%, hoger bij kinderen. De doodsoorzaken bij volwassenen zijn ernstige en progressieve spierzwakte, dysfagie, verminderde voeding, aspiratie-pneumonie of ademhalingsinsufficiëntie als gevolg van longinfecties. Poliomyositis is ernstiger en resistenter tegen behandeling in aanwezigheid van beschadiging van het hart en de longen. Dood bij kinderen kan optreden als gevolg van intestinale vasculitis. De algehele prognose van de ziekte wordt ook bepaald door de aanwezigheid van maligne neoplasma's.