Medisch expert van het artikel

Nieuwe publicaties
Subacute necrotiserende Leah's encefalomyopathie
Laatst beoordeeld: 04.07.2025

Alle iLive-inhoud wordt medisch beoordeeld of gecontroleerd op feiten om zo veel mogelijk feitelijke nauwkeurigheid te waarborgen.
We hebben strikte richtlijnen voor sourcing en koppelen alleen aan gerenommeerde mediasites, academische onderzoeksinstellingen en, waar mogelijk, medisch getoetste onderzoeken. Merk op dat de nummers tussen haakjes ([1], [2], etc.) klikbare links naar deze studies zijn.
Als u van mening bent dat onze inhoud onjuist, verouderd of anderszins twijfelachtig is, selecteert u deze en drukt u op Ctrl + Enter.
De ziekte werd voor het eerst genoemd in 1951. Tot op heden zijn er meer dan 120 gevallen beschreven. De ziekte van Leigh (OMIM 256000) is een genetisch heterogene ziekte die zowel nucleair (autosomaal recessief of X-gebonden) als mitochondriaal (minder vaak) kan worden overgeërfd.
 [
[ Oorzaken van het syndroom van Leah
De ziekte is gebaseerd op een tekort aan enzymen die zorgen voor energieproductie, voornamelijk veroorzaakt door een verstoring van het pyrodruivenzuurmetabolisme en een defect in het elektronentransport in de ademhalingsketen. Er ontstaat een tekort aan het pyruvaatdehydrogenasecomplex (a-E1-subeenheid), pyruvaatcarboxylase, complex 1 (NAD-co-enzym Q-reductase) en complex 4 (cytochroomoxidase) van de ademhalingsketen.
Het is vastgesteld dat defecten van pyruvaatcarboxylase, complex 1 (NAD-co-enzym Q-reductase) en complex 4 (cytochroomoxidase) van de ademhalingsketen autosomaal recessief overerven, terwijl defecten van het pyruvaatdehydrogenasecomplex (a-E1-subeenheid) X-gebonden recessief overerven. Bij puntmutaties van het mtDNA, die de 6e subeenheid van ATPase beïnvloeden, is mitochondriale overerving kenmerkend. Meestal treedt een miscens-mutatie op, geassocieerd met de vervanging van thymine door guanine of cytosine op positie 8993 van het mtDNA. Minder vaak komt een mutatie op positie 9176 van het mtDNA voor. Omdat de T8993G-mutatie het belangrijkste defect is bij het NARP-syndroom, zijn er families met deze twee aandoeningen beschreven. Bij kinderen is ook een mutatie in het mtDNA op positie 8344 beschreven, die voorkomt bij het MERRF-syndroom.
Aangenomen wordt dat bij ophoping van gemuteerd mtDNA in de meeste mitochondriën een ernstig beloop van het syndroom van Leigh ontstaat. Bij de mitochondriale oorsprong van deze aandoening wordt gemuteerd mtDNA in 90% van alle mitochondriën aangetroffen. De pathogenese gaat gepaard met een verstoring van de energieproductie in cellen en de ontwikkeling van lactaatacidose.
Symptomen van het syndroom van Leah
De eerste tekenen van de ziekte openbaren zich op jonge leeftijd (1-3 jaar). Er zijn echter gevallen bekend van de ziektemanifestatie op 2 weken en op 6-7 jaar. Aanvankelijk ontwikkelen zich niet-specifieke stoornissen: vertraagde psychomotorische ontwikkeling, verminderde eetlust, braken, gewichtsverlies. Vervolgens nemen de neurologische symptomen toe: spierhypotonie of dystonie met overgang naar hypertonie, aanvallen van myoclonus of tonisch-clonische aanvallen, tremor van de extremiteiten, choreoathetose, coördinatiestoornis, verminderde peesreflexen, lethargie, slaperigheid. Cerebrale neurodegeneratie is progressief. Symptomen van piramidale en extrapiramidale insufficiëntie nemen toe, het slikken wordt belemmerd. Veranderingen in het gezichtsorgaan zoals ptosis, oftalmoplegie, atrofie van de oogzenuwen, minder vaak pigmentaire degeneratie van het netvlies, worden vaak waargenomen. Soms ontwikkelt zich hypertrofische cardiomyopathie, episodes van tachypneu.
Zelden verloopt de ziekte als een acute encefalopathie. Meer kenmerkend is een chronisch of subacuut beloop, dat enkele jaren na het begin van de ziekte fataal afloopt. Bij een snel beloop (enkele weken) treedt de dood in als gevolg van verlamming van het ademhalingscentrum.
Diagnostics van het syndroom van Leah
Een biochemische bloedtest toont lactaatacidose aan als gevolg van de ophoping van melkzuur en pyrodruivenzuur in het bloed en het hersenvocht, evenals een verhoogd alaninegehalte in het bloed. Het gehalte aan ketonlichamen kan ook verhoogd zijn. Een verhoogde uitscheiding van organische zuren in de urine wordt vastgesteld: melkzuur, fumaarzuur, enz. Het carnitinegehalte in het bloed en de weefsels daalt vaak.
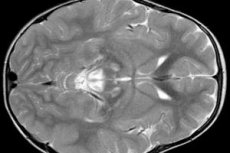
EEG-resultaten tonen focale tekenen van epileptische activiteit. MRI-gegevens tonen vergroting van de hersenventrikels, bilaterale hersenschade en verkalking van de basale ganglia (nucleus caudatus, putamen, substantia nigra, globus pallidus). Atrofie van de hersenhelften en hersenweefsel kan ook worden vastgesteld.
Morfologisch onderzoek toont duidelijke veranderingen in de hersenmaterie aan: symmetrische necrotische foci, demyelinisatie en sponsachtige degeneratie van de hersenen, voornamelijk van de middelste hersenhelften, de pons, de basale ganglia, de thalamus en de oogzenuw. Het histologische beeld omvat cystische degeneratie van hersenweefsel, astrocytaire gliose, neuronale afsterving en een toename van het aantal mitochondriën in de cellen. In de skeletspieren is er sprake van accumulatie van lipide-insluitsels, een afname van de histochemische reactie op complex 1 en 4 van de ademhalingsketen, subsarcolemmale accumulatie van mitochondriën, en abnormale mitochondriën met desorganisatie van de cristae. Het RRF-fenomeen wordt vaak niet gedetecteerd.
Hoe te onderzoeken?
Welke tests zijn nodig?
Использованная литература