Medisch expert van het artikel

Nieuwe publicaties
Alcaptonurie - aangeboren enzymatische pathologie
Laatst beoordeeld: 23.04.2024

Alle iLive-inhoud wordt medisch beoordeeld of gecontroleerd op feiten om zo veel mogelijk feitelijke nauwkeurigheid te waarborgen.
We hebben strikte richtlijnen voor sourcing en koppelen alleen aan gerenommeerde mediasites, academische onderzoeksinstellingen en, waar mogelijk, medisch getoetste onderzoeken. Merk op dat de nummers tussen haakjes ([1], [2], etc.) klikbare links naar deze studies zijn.
Als u van mening bent dat onze inhoud onjuist, verouderd of anderszins twijfelachtig is, selecteert u deze en drukt u op Ctrl + Enter.
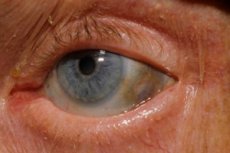
Een van de zeer zeldzame stofwisselingsstoornissen - alkaptonurie - verwijst naar aangeboren afwijkingen in het metabolisme van het aminozuur tyrosine.
Dit syndroom kan ook homogentisaatoxidasedeficiëntie, homogentisinurie, erfelijke ochronose of zwarte urineziekte worden genoemd. [1]
Epidemiologie
Volgens statistieken zijn er niet meer dan negen gevallen van alkaptonurie per 1 miljoen van de bevolking. En in de meeste Europese landen - één geval per 100-250 duizend levendgeborenen.
Van de Europese landen is de uitzondering Slowakije (vooral de relatief kleine noordwestelijke regio), waar de prevalentie van alkaptonurie één geval is op 19 duizend pasgeborenen. Naar alle waarschijnlijkheid is dit te wijten aan het feit dat onder de families van Slowaakse Roma die daar wonen, het niveau van inteelt (huwelijken tussen neven en nichten) het hoogste is in Europa: 10-14%. [2]
Oorzaken alkaptonurie
De exacte oorzaken van alkaptonurie, als een aangeboren stoornis van katabolisme (metabole afbraak) van het aromatische (homocyclische) α-aminozuur tyrosine, zijn vastgesteld: dit type stofwisselingsstoornis is een gevolg van homozygote of complexe heterozygote mutaties van één op de duizenden van genen op chromosoom 3, meer bepaald het HqG21-gen op locus 3-q23 op de lange arm van het chromosoom. Dit gen codeert voor de nucleotidesequenties van het leverenzym homogentisaat-1,2-dioxygenase [3](ook wel homogentisinezuuroxidase of homogentisaatoxidase genoemd), een ijzerhoudend metalloproteïne dat nodig is voor een van de stadia van tyrosineafbraak in het lichaam. [4], [5]
Alkaptonurie is dus een defect in het enzym homogentisaat-1,2-dioxygenase, meer bepaald het resultaat van zijn genetisch bepaalde deficiëntie of volledige afwezigheid. [6]
Omdat het een congenitale fermentopathie is, wordt alkaptonurie geërfd als een autosomaal recessieve eigenschap, dat wil zeggen dat om alkaptonurie bij kinderen te laten optreden, beide ouders een gemodificeerd gen voor dit enzym moeten hebben, aangezien elk van hen slechts één kopie van de gen van de twee beschikbare.
Volgens de laatste gegevens zijn er meer dan tweehonderd varianten van de HGD-genmodificatie en worden missense-mutaties, translocatie en splicing het vaakst waargenomen.
Risicofactoren
De enige risicofactor voor de ontwikkeling van deze aangeboren fermentopathie is de familiegeschiedenis en de overerving van twee gemodificeerde kopieën van het HGD-gen, als de ouders geen alkaptonurie vertonen (het risico van overdracht van de afwijking is 25%), of een van de de ouders hebben deze aandoening. [7]
Pathogenese
De belangrijkste rol die tyrosine speelt bij de synthese van eiwitten, de productie van chromoproteïnen - het huidpigment melanine, evenals schildklierhormonen en catecholamine-neurotransmitters.
Het mechanisme voor het reguleren van de hoeveelheid tyrosine in cellen is zeer complex en het lichaam normaliseert het overtollige gehalte door te splitsen. Het proces van katabolisme van tyrosine, zoals alle aromatische aminozuren, is meerstaps en verloopt in verschillende fasen. Bovendien vindt elk stadium van de metabolische afbraak van tyrosine plaats met de deelname van een bepaald enzym en de vorming van een tussenproduct.
Dus eerst wordt het aminozuur gesplitst tot para-hydroxyfenylpyruvaat, dat verandert in een alkapon - 2,5-dihydroxyfenylazijnzuur of homogentisinezuur. Verder moet het alkapon worden omgezet in maleazijnzuur, maar dit gebeurt niet. [8]
En de pathogenese van alkaptonurie bestaat uit het stoppen van de biochemische reacties van tyrosinekatabolisme in het stadium van vorming van homogentisinezuur: er is eenvoudigweg geen noodzakelijk enzym om het af te breken - homogentisaatoxidase.
Homogentisinezuur wordt niet door het lichaam gebruikt en kan zich ophopen bij uitscheiding via de nieren. Bovendien wordt het geoxideerd tot benzoquinoacetaat (benzoquinonazijnzuur), dat door binding met moleculen van weefsels en lichaamsvloeistoffen biopolymeerverbindingen vormt die gekleurd zijn als melanine.
De ophoping van deze tussenproducten in het weefsel leidt tot verstoring van de collageenstructuur van het kraakbeenweefsel, waardoor de elasticiteit ervan afneemt - met het optreden van veel klinische symptomen van alkaptonurie en de ontwikkeling van complicaties.
Symptomen alkaptonurie
Alcaptonurie bij pasgeborenen en zuigelingen manifesteert zich door donker worden van de urine. Bij contact met lucht wordt de kleur van urine op luiers, luiers en ondergoed donkerbruin; dit komt door de ophoping en afgifte van homogentisinezuur, dat wordt geoxideerd tot benzoquinoacetaat. [9]
Bij afwezigheid van andere symptomen wordt alkaptonurie bij kinderen op jonge leeftijd vaak niet tijdig herkend, omdat na het plassen de urine na enkele uren donker kan worden. Volgens sommige rapporten wordt in de omstandigheden van klinieken slechts een vijfde van de kinderen jonger dan 12 maanden die met deze fermentopathie zijn geboren, gedetecteerd. Daarom is het erg belangrijk dat ouders aandacht besteden aan de zorg voor de baby.
Bovendien omvatten vroege tekenen pigmentatie (blauwgrijs) van de sclera van het oog en het kraakbeen van de oorschelpen en neus, vaak aangeduid als ochronosis. [10]
Na verloop van tijd verschijnen er andere symptomen:
- ernstige pigmentatie van de huid op de jukbeenderen, oksels en geslachtsdelen;
- kleding verven bij contact met zwetende lichaamsdelen;
- aanvallen van algemene zwakte;
- Schorre stem.
Houd er rekening mee dat alkaptonurie en ochronose , zoals hierboven vermeld, synoniemen zijn voor dezelfde schending van tyrosinekatabolisme.
Ahornsiroopziekte en alkaptonurie. Congenitale ahornsiroopziekte of leucinose verwijst ook naar stofwisselingsstoornissen, heeft hetzelfde type overerving en zelfs mutaties komen voor op hetzelfde chromosoom, maar hebben betrekking op het gen dat codeert voor het enzymcomplex van vertakte α-ketozuren dehydrogenase. Hierdoor kan het lichaam bepaalde eiwitcomponenten niet afbreken, met name de aminozuren leucine, isoleucine en valine. In deze toestand heeft urine (en oorsmeer) een zoete geur; bovendien worden in het klinische beeld van dit type organische acidemie hypopigmentatie, fluctuaties in bloeddruk, convulsies, braken en diarree, een daling van de bloedglucosespiegels, ketoacidose, hallucinaties, enz. Waargenomen.Het sterftecijfer bij kinderen is vrij hoog; bij volwassenen zonder behandeling kunnen coma en overlijden optreden als gevolg van hersenoedeem.
Albinisme en alkaptonurie "verenigen" alleen tyrosine. Albinisme , inclusief oculocutaan albinisme, wordt veroorzaakt door genetische mutaties die de productie van het pigment melanine beïnvloeden. Aangeboren veranderingen worden opgemerkt in het TYR-gen op chromosoom 11 (11q14.3), dat codeert voor tyrosinase, een koperbevattend enzym in melanosomen dat nodig is voor de vorming van huidpigment op basis van de producten van het tyrosinemetabolisme. Deze ziekte komt veel vaker voor dan alkaptonurie.
Complicaties en gevolgen
De gevolgen en complicaties van alkaptonurie veroorzaakt door de werking van de intermediaire tyrosinemetabolieten - homogentisische en benzochinonazijnzuren - verschijnen als gevolg van de afzetting van reactieve gepigmenteerde polymeren, de vernietiging van collageenfibrillen en de verslechtering van de kraakbeenconditie (met een afname van hun weerstand mechanische belasting).
Jaren later, op volwassen leeftijd, ontwikkelen zich degeneratieve artritis en artrose van grote gewrichten (heup, sacro-iliacale en knie); vernauwing van de tussenwervelruimten (vooral van de lumbale en thoracale wervelkolom) - met verkalking en de vorming van osteofyten; de weefseldichtheid van de subchondrale botplaten neemt af en de onderliggende botten kunnen pathologische hermodellering ondergaan met de vorming van gezwellen en vervorming. [11]
Schade aan de hartkleppen (aorta en mitralis) en kransslagaders, met tekenen van coronaire hartziekte, evenals de vorming van nier- en prostaatstenen, als gevolg van dezelfde verkalking, kan optreden. [12], [13]
Diagnostics alkaptonurie
Meestal is de diagnose van aangeboren stofwisselingsstoornissen gebaseerd op de studie van lichaamsvloeistoffen.
Op basis van welke tests en welke reacties kan alkaptonurie worden gediagnosticeerd? Urinetests zijn vereist - voor het detecteren van homogentisinezuur en het bepalen van het niveau (normaal - 20-30 mg per dag, verhoogd - 3-8 g). Een urinemonster wordt onderzocht door middel van gaschromatografie of massaspectrometrie met behulp van vloeistofchromatografie; een screeningstest op de aanwezigheid van ijzerchloride in de urine is mogelijk. [14]
Er is ook een snelle diagnostische methode - bepaling van alkapton in gedroogde urinevlekken op papier (op kleurintensiteit).
Bij het verduidelijken van de diagnose omvat instrumentele diagnostiek (röntgenfoto) de identificatie van röntgensignalen van artrose en andere gewrichtspathologieën bij patiënten.
De diagnose wordt bevestigd door& moleculair genetische methoden zoals diagnostiek van erfelijke ziekten , zoals genetische testen en DNA-sequencing. [15]
Differentiële diagnose
Differentiële diagnose Differentiële diagnoses omvatten hemochromatose en acuut leverfalen bij pasgeborenen, melaninurie, acute intermitterende porfyrie, hemofagocytische lymfohistiocytose, primaire mitochondriale pathologie, reumatoïde artritis, spondylitis ankylopoetica.
Met wie kun je contact opnemen?
Behandeling alkaptonurie
De belangrijkste behandeling voor alkaptonurie is de inname van grote doses (minimaal 1000 mg per dag) ascorbinezuur. Bij kinderen verhoogt dit de uitscheiding van homogentisinezuur in de urine, en bij volwassenen verlaagt het het niveau van zijn derivaat, benzochinonazijnzuur, in de urine en vertraagt het de binding aan de bindweefselstructuren van de gewrichten en collageen. [16]
In West-Europese klinieken wordt het medicijn Nitizinone (Orfalin) getest, een medicijn van een groep metabolieten die de tweede fase van tyrosinekatabolisme remt: de transformatie van para-hydroxyfenylpyruvaat in homogentisinezuur. Het gebruik van dit farmacologische middel leidt echter tot de ophoping van tyrosine en kan ernstige bijwerkingen veroorzaken, waaronder troebelheid van het hoornvlies en fotofobie, neus- en maagbloedingen, leverfalen, veranderingen in het bloed, enz. In de Verenigde Staten wordt Nithizinon echter is goedgekeurd door de FDA voor de behandeling van tyrosinemie I type. [17], [18]
Zo wordt fysiotherapie - oefentherapie om spierkracht te vergroten en gewrichtsmobiliteit te vergroten, balneo- en peloidotherapie om pijn te beperken - uitgevoerd bij gewrichtsproblemen veroorzaakt door alkaptonurie.
Hoewel tyrosine niet alleen met voedsel wordt geleverd, maar ook door het lichaam wordt geproduceerd, wordt patiënten met alkaptonurie aanbevolen een eiwitarm dieet te volgen en de inname van voedsel dat rijk is aan tyrosine te beperken, voornamelijk rundvlees en varkensvlees, zuivelproducten (vooral kazen), peulvruchten, noten, enz. Zaden.
Het voorkomen
Preventie van genmutaties is onmogelijk, en om de geboorte van kinderen met een hoog risico op aangeboren aandoeningen te voorkomen, is er medische en genetische counseling, die nodig is vóór de geplande zwangerschap van die echtparen in wiens familiegeschiedenis ziekten van een erfelijke natuur. [19]
Prognose
Er zijn zeer weinig dodelijke slachtoffers in de loop van alkaptonurie, en de dood kan worden veroorzaakt door ernstige complicaties die het hart en de nieren aantasten. Dus voor de totale levensverwachting van mensen met alkaptonurie is de prognose gunstig.
Maar de kwaliteit van leven wordt verminderd door intense pijn in de gewrichten of de wervelkolom met een aanzienlijke beperking van de mobiliteit, vaak progressief.