Medisch expert van het artikel

Nieuwe publicaties
Subacute necrotiserende encefalomyopathie Leia
Laatst beoordeeld: 23.04.2024

Alle iLive-inhoud wordt medisch beoordeeld of gecontroleerd op feiten om zo veel mogelijk feitelijke nauwkeurigheid te waarborgen.
We hebben strikte richtlijnen voor sourcing en koppelen alleen aan gerenommeerde mediasites, academische onderzoeksinstellingen en, waar mogelijk, medisch getoetste onderzoeken. Merk op dat de nummers tussen haakjes ([1], [2], etc.) klikbare links naar deze studies zijn.
Als u van mening bent dat onze inhoud onjuist, verouderd of anderszins twijfelachtig is, selecteert u deze en drukt u op Ctrl + Enter.
 [
[Oorzaken van het Leia-syndroom
De kern van de ziekte is een tekort aan enzymen die zorgen voor energie, voornamelijk als gevolg van een verstoring van het metabolisme van pyrodruivenzuur en een defect in het transport van elektronen in de ademhalingsketen. Pyruvaat dehydrogenase complex deficiëntie ontwikkelt (a-subunit E1), pyruvaat carboxylase, complex 1 (NAD coenzym Q-reductase) en complexe 4 (cytochroom oxidase) de ademhalingsketen.
Gevonden werd dat defecten pyruvaat, complex 1 (NAD coenzym Q-reductase) en complexe 4 (cytochroom oxidase) de ademhalingsketen worden autosomaal recessieve wijze, defecten van het pyruvaat dehydrogenase complex (a-E1 subeenheid) - X-gebonden recessieve. Wanneer mtDNA puntmutaties die invloed hebben op 6-ATPase subunit, mitochondriale overerving karakteristiek. Meestal gebeurt mistsens mutatie in verband met de vervanging van thymine naar guanine of cytosine op positie 8993 mtDNA. Minder gebruikelijk is een mutatie op positie 9176 mtDNA. Vanwege het feit dat mutaties T8993G - basisdefect het syndroom NARP in de familie beschreven aan de aanwezigheid van deze twee ziekten. Kinderen zijn ook beschreven mtDNA mutatie op positie 8344, die is gevonden in het syndroom MERRF.
Er wordt gesuggereerd dat in het geval van accumulatie van mutant mtDNA in de meerderheid van mitochondriën een ernstige ontwikkeling van het Leia-syndroom optreedt. In de mitochondriale genese van deze toestand wordt mutant mtDNA gedetecteerd in 90% van alle mitochondriën. Pathogenese gaat gepaard met een schending van de energieproductie in cellen en de ontwikkeling van melkzuuracidose.
Symptomen van het Leia-syndroom
De eerste tekenen van het ziektedebuut op jonge leeftijd (1-3 jaar). Echter, er zijn gevallen van de ziekte manifestaties in de 2-week en op 6-7 jaar. In eerste instantie ontwikkeld non-specifieke aandoeningen: psychomotorische retardatie, verlies van eetlust, braken afleveringen, ondergewicht. In de daaropvolgende groeiende neurologische symptomen: spierhypotonie of dystonie bij de overgang naar hypertonie, convulsies, myoclonische schokken of tonisch-klonische aanvallen, tremor ledematen, choreoathetose, coördinatie stoornis, verminderde peesreflexen, lethargie, slaperigheid. Cerebrale neurodegeneratie heeft een progressief karakter. Het oppakken van de symptomen van de piramidale en extrapiramidale ziekte, slikstoornissen. Vaak is er een dergelijke verandering van gezag als ptosis, ophthalmoplegia, optische atrofie, retinitis pigmentosa minder. Soms ontwikkelen hypertrofische cardiomyopathie, zijn er afleveringen van tachypneu.
In zeldzame gevallen verloopt de ziekte volgens het type acute encefalopathie. Meer karakteristiek is een chronische of subacute stroom, die enkele jaren na het begin van de ziekte tot een fatale afloop leidt. Bij een snelle stroom (enkele weken) treedt de dood op als gevolg van verlamming van het ademhalingscentrum.
Diagnostics van het Leia-syndroom
In een biochemische bloedtest wordt lactaatacidose gedetecteerd als gevolg van de ophoping van melkzuur en pyrodruivenzuren in het bloed en de liquor, evenals een toename van het gehalte aan alanine in het bloed. Ook kan het niveau van ketonlichamen worden verhoogd. In de urine is er een verhoogde uitscheiding van organische zuren: melkzuur, fumaarzuur, enz. Het niveau van carnitine in het bloed en de weefsels wordt vaak verminderd.
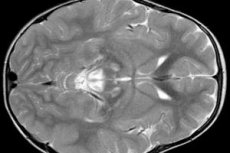
EEG-resultaten onthullen focale tekenen van epileptische activiteit. Volgens MRI-gegevens worden een uitbreiding van de ventrikels van de hersenen, bilaterale hersenschade, verkalking van de basale ganglia (caudate nucleus, schaal, zwarte substantie, blinde bol) gedetecteerd. Het is ook mogelijk om atrofie van de hersenhelften en hersenstoffen te identificeren.
Morfologische studie tonen grote veranderingen in hersenmaterie: symmetrische necrose, demyelinisatie en sponsachtige degeneratie van de hersenen, vooral middendelen, brug, basale ganglia, thalamus, oogzenuw. Het histologische beeld omvat cystische degeneratie van het hersenweefsel, astrocytische gliosis, dood van neuronen, een toename van het aantal mitochondriën in de cellen. In skeletspier - de ophoping van lipide insluitsels verminderen histochemische reactie van 1-, 4 ademhalingsketen van mitochondriën subsarkolemmalnoe congestie, abnormale mitochondria verstoring van cristae. Het fenomeen van RRF wordt vaak niet gedetecteerd.
Hoe te onderzoeken?
Welke tests zijn nodig?
Использованная литература