Medisch expert van het artikel

Nieuwe publicaties
Erfelijke nefritis (Alport-syndroom) bij kinderen
Laatst beoordeeld: 05.07.2025

Alle iLive-inhoud wordt medisch beoordeeld of gecontroleerd op feiten om zo veel mogelijk feitelijke nauwkeurigheid te waarborgen.
We hebben strikte richtlijnen voor sourcing en koppelen alleen aan gerenommeerde mediasites, academische onderzoeksinstellingen en, waar mogelijk, medisch getoetste onderzoeken. Merk op dat de nummers tussen haakjes ([1], [2], etc.) klikbare links naar deze studies zijn.
Als u van mening bent dat onze inhoud onjuist, verouderd of anderszins twijfelachtig is, selecteert u deze en drukt u op Ctrl + Enter.
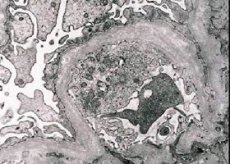
Erfelijke nefritis (syndroom van Alport) is een genetisch bepaalde, erfelijke, niet-immuun glomerulopathie die zich manifesteert door hematurie (soms met proteïnurie), progressieve achteruitgang van de nierfunctie met de ontwikkeling van chronisch nierfalen, vaak gecombineerd met sensorineurale doofheid en slechtziendheid.
De ziekte werd voor het eerst beschreven in 1902 door LG Guthrie, die een familie observeerde waarin hematurie in meerdere generaties voorkwam. In 1915 beschreef AF Hurst de ontwikkeling van uremie bij leden van dezelfde familie. In 1927 identificeerde A. Alport voor het eerst gehoorverlies bij verschillende familieleden met hematurie. In de jaren vijftig werden oogafwijkingen bij een vergelijkbare ziekte beschreven. In 1972 ontdekten Hinglais et al. tijdens een morfologisch onderzoek van nierweefsel bij patiënten met erfelijke hematurie een ongelijkmatige expansie en stratificatie van de glomerulaire basaalmembranen. In 1985 werd de genetische basis van erfelijke nefritis geïdentificeerd: een mutatie in het gen voor collageen type IV (Fiengold et al., 1985).
De studie van de genetische aard van de ziekte bracht ons tot de conclusie dat de verschillen in de fenotypische manifestaties van erfelijke nefritis (met of zonder gehoorverlies) te wijten zijn aan de mate van expressie van het gemuteerde gen. Daarom worden momenteel alle klinische varianten beschouwd als manifestaties van één ziekte en is de term "erfelijke nefritis" synoniem met de term "syndroom van Alport".
Volgens epidemiologische studies komt erfelijke nefritis voor met een frequentie van 17 per 100.000 kinderen.
 [
[ Oorzaken van het Alport-syndroom
De genetische basis van de ziekte is een mutatie in het gen van de a-5-keten van type IV collageen. Dit type is universeel voor de basale membranen van de nieren, het cochlea-apparaat, het lenskapsel, het netvlies en het hoornvlies van het oog, wat is aangetoond in studies met monoklonale antilichamen tegen deze collageenfractie. Recent is de mogelijkheid van het gebruik van DNA-probes voor prenatale diagnostiek van erfelijke nefritis geïndiceerd.
Het belang van het testen van alle familieleden met DNA-sondes om dragers van het gemuteerde gen te identificeren, wordt benadrukt. Dit is van groot belang bij de medische en genetische begeleiding van families met deze ziekte. Tot 20% van de families heeft echter geen familieleden die aan een nieraandoening lijden, wat wijst op een hoge frequentie van spontane mutaties van het afwijkende gen. De meeste patiënten met erfelijke nefritis hebben personen met een nieraandoening, gehoorverlies en visuele pathologie in hun familie; consanguine huwelijken tussen personen met een of meer voorouders zijn belangrijk, omdat bij een huwelijk tussen verwante personen de kans toeneemt dat ze dezelfde genen van beide ouders ontvangen. Er zijn autosomaal dominante, autosomaal recessieve en dominante, X-gebonden transmissieroutes vastgesteld.
Bij kinderen worden de drie meest voorkomende vormen van erfelijke nefritis onderscheiden: het syndroom van Alport, erfelijke nefritis zonder gehoorverlies en familiaire goedaardige hematurie.
Het syndroom van Alport is een erfelijke nefritis met gehoorverlies. Het is gebaseerd op een gecombineerd defect in de structuur van het collageen van de glomerulaire basaalmembraan van de nieren, oren en ogen. Het gen voor het klassieke syndroom van Alport bevindt zich in locus 21-22 q van de lange arm van het X-chromosoom. In de meeste gevallen wordt het dominant overgeërfd, gekoppeld aan het X-chromosoom. In dit opzicht is het syndroom van Alport ernstiger bij mannen, omdat bij vrouwen de functie van het gemuteerde gen wordt gecompenseerd door een gezond allel van het tweede, onbeschadigde chromosoom.
De genetische basis voor het ontstaan van erfelijke nefritis wordt gevormd door mutaties in de genen van de alfaketens van type IV collageen. Er zijn zes alfaketens van type IV collageen G bekend: de genen van de a5- en a6-ketens (Col4A5 en Col4A5) bevinden zich op de lange arm van het X-chromosoom in de zone 21-22q; de genen van de a3- en a4-ketens (Col4A3 en Col4A4) bevinden zich op het 2e chromosoom; de genen van de a1- en a2-ketens (Col4A1 en Col4A2) bevinden zich op het 13e chromosoom.
In de meeste gevallen (80-85%) wordt een X-gebonden overervingspatroon van de ziekte vastgesteld, geassocieerd met schade aan het Col4A5-gen als gevolg van deletie, puntmutaties of splicingstoornissen. Momenteel zijn er meer dan 200 mutaties van het Col4A5-gen gevonden, die verantwoordelijk zijn voor de verstoring van de synthese van de a5-ketens van type IV collageen. Bij dit type overerving manifesteert de ziekte zich bij kinderen van beide geslachten, maar bij jongens is de ziekte ernstiger.
Mutaties in de loci van de genen Col4A3 en Col4A4, die verantwoordelijk zijn voor de synthese van de a3- en a4-ketens van type IV collageen, worden autosomaal overgeërfd. Volgens onderzoek wordt de autosomaal dominante overerving waargenomen bij 16% van de gevallen van erfelijke nefritis en de autosomaal recessieve overerving bij 6% van de patiënten. Er zijn ongeveer 10 varianten van mutaties van de genen Col4A3 en Col4A4 bekend.
Mutaties resulteren in een verstoring van de assemblageprocessen van collageen type IV, wat leidt tot een verstoring van de structuur ervan. Collageen type IV is een van de belangrijkste componenten van het glomerulaire basaalmembraan, het cochlea-apparaat en de ooglens. De pathologie hiervan wordt vastgesteld in de kliniek voor erfelijke nefritis.
Collageen type IV, dat deel uitmaakt van de glomerulaire basaalmembraan, bestaat voornamelijk uit twee a1-ketens (IV) en één a2-keten (IV), en bevat ook a3-, a4- en a5-ketens. Bij X-gebonden overerving gaat de mutatie van het Col4A5-gen meestal gepaard met de afwezigheid van a3-, a4-, a5- en a6-ketens in de structuur van collageen type IV, en neemt het aantal o1- en a2-ketens in de glomerulaire basaalmembraan toe. Het mechanisme van dit fenomeen is onduidelijk; men neemt aan dat de oorzaak ligt in posttranscriptionele veranderingen in mRNA.
De afwezigheid van a3-, a4- en a5-ketens in de structuur van collageen type IV van de glomerulaire basale membranen leidt tot verdunning en fragiliteit ervan in de vroege stadia van het syndroom van Alport, wat zich klinisch vaker manifesteert door hematurie (minder vaak door hematurie met proteïnurie of alleen proteïnurie), gehoorverlies en lenticonus. Verdere progressie van de ziekte leidt tot verdikking en verminderde permeabiliteit van de basale membranen in de late stadia van de ziekte, met de proliferatie van collageen type V en VI daarin, wat zich manifesteert in een toename van proteïnurie en een afname van de nierfunctie.
De aard van de mutatie die ten grondslag ligt aan erfelijke nefritis bepaalt grotendeels de fenotypische manifestatie ervan. Bij een deletie van het X-chromosoom met gelijktijdige mutatie van de Col4A5- en Col4A6-genen, verantwoordelijk voor de synthese van de a5- en a6-ketens van type IV collageen, gaat het syndroom van Alport gepaard met leiomyomatose van de slokdarm en de geslachtsdelen. Volgens onderzoeksgegevens wordt bij een mutatie van het Col4A5-gen geassocieerd met een deletie een ernstigere pathologische ontwikkeling waargenomen, een combinatie van nierschade met extrarenale manifestaties en een vroege ontwikkeling van chronisch nierfalen, vergeleken met een puntmutatie van dit gen.
Morfologisch gezien onthult elektronenmicroscopie verdunning en stratificatie van de glomerulaire basale membranen (met name de lamina densa) en de aanwezigheid van elektronendichte granula. Glomerulaire laesies kunnen heterogeen zijn bij dezelfde patiënt, van minimale focale mesangiale laesies tot glomerulosclerose. Glomerulitis bij het syndroom van Alport is altijd immunonegatief, wat het onderscheidt van glomerulonefritis. Karakteristieke kenmerken zijn onder meer de ontwikkeling van tubulaire atrofie, lymfohistiocytaire infiltratie en de aanwezigheid van "schuimcellen" met lipide-insluitsels - lipofagen. Naarmate de ziekte vordert, worden verdikking en uitgesproken vernietiging van de glomerulaire basale membranen zichtbaar.
Er worden bepaalde veranderingen in het immuunsysteem zichtbaar. Patiënten met erfelijke nefritis hebben een verlaagde IgA-spiegel en een neiging tot een verhoogde IgM-concentratie in het bloed. De IgG-spiegel kan in de vroege stadia van de ziekte verhoogd zijn en in de latere stadia afnemen. Mogelijk is de verhoogde IgM- en G-concentratie een soort compensatiereactie op een IgA-tekort.
De functionele activiteit van het T-lymfocytensysteem is verminderd; er wordt een selectieve afname opgemerkt van B-lymfocyten die verantwoordelijk zijn voor de synthese van Ig A, de fagocytische link van de immuniteit is verstoord, voornamelijk als gevolg van verstoring van chemotaxis en intracellulaire verteringsprocessen in neutrofielen
Bij onderzoek van een nierbiopsie bij patiënten met het syndroom van Alport laten elektronenmicroscopiegegevens ultrastructurele veranderingen in de glomerulaire basaalmembraan zien: verdunning, verstoring van de structuur en splitsing van de glomerulaire basaalmembranen met een verandering in de dikte en onregelmatige contouren. In de vroege stadia van erfelijke nefritis bepaalt het defect de verdunning en kwetsbaarheid van de glomerulaire basaalmembranen.
Verdunning van de glomerulaire membranen is een gunstiger teken en komt vaker voor bij meisjes. Een constanter elektronenmicroscopisch teken bij erfelijke nefritis is de scheuring van de basale membraan, en de ernst van de vernietiging correleert met de ernst van het proces.
Symptomen van het Alport-syndroom bij kinderen
De eerste symptomen van het syndroom van Alport, in de vorm van een geïsoleerd urinair syndroom, worden meestal ontdekt bij kinderen in de eerste drie levensjaren. In de meeste gevallen wordt de ziekte bij toeval ontdekt. Het urinair syndroom wordt ontdekt tijdens een preventief onderzoek van het kind, vóór opname in een kinderdagverblijf of tijdens een acute respiratoire insufficiëntie (SARS). Bij een SARS-infectie is er bij een SARS-infectie sprake van pathologie in de urine. Bij erfelijke nefritis is er, in tegenstelling tot verworven glomerulonefritis, geen latente periode.
In het beginstadium van de ziekte lijdt de gezondheid van het kind er weinig onder; een kenmerkend kenmerk is de persistentie en resistentie van het urinaire syndroom. Een van de belangrijkste symptomen is hematurie van verschillende ernst, die in 100% van de gevallen wordt waargenomen. Een toename van de mate van hematurie wordt opgemerkt tijdens of na luchtweginfecties, fysieke activiteit of na preventieve vaccinaties. Proteïnurie bedraagt in de meeste gevallen niet meer dan 1 g/dag; aan het begin van de ziekte kan de proteïnurie onregelmatig zijn, maar naarmate het proces vordert, neemt de proteïnurie toe. Regelmatig kan leukocyturie met een overwicht aan lymfocyten in het urinesediment voorkomen, wat gepaard gaat met de ontwikkeling van interstitiële veranderingen.
Vervolgens is de nierfunctie gedeeltelijk verminderd en verslechtert de algemene toestand van de patiënt: intoxicatie, spierzwakte, arteriële hypotensie, vaak gehoorverlies (vooral bij jongens) en soms ook visuele stoornissen. Intoxicatie manifesteert zich door bleekheid, vermoeidheid en hoofdpijn. In het beginstadium van de ziekte wordt gehoorverlies in de meeste gevallen alleen vastgesteld door middel van audiografie. Gehoorverlies bij het syndroom van Alport kan op verschillende momenten in de kindertijd optreden, maar meestal wordt gehoorverlies vastgesteld op de leeftijd van 6-10 jaar. Gehoorverlies bij kinderen begint met hoge frequenties en bereikt een significante mate van lucht- en botgeleiding, waarbij het overgaat van geluidgeleidend naar geluidwaarnemend gehoorverlies. Gehoorverlies kan een van de eerste symptomen van de ziekte zijn en kan voorafgaan aan het urinair syndroom.
In 20% van de gevallen hebben patiënten met het syndroom van Alport veranderingen in de gezichtsorganen. De meest frequent vastgestelde afwijkingen zijn die van de lens: sferofokie, anterieure, posterieure of gemengde lenticonus en diverse cataracten. In families met het syndroom van Alport komt myopie significant voor. Een aantal onderzoekers merkt in deze families voortdurend bilaterale perimaculaire veranderingen op in de vorm van heldere witachtige of gelige granulaties in het corpus luteum. Zij beschouwen dit teken als een constant symptoom met een hoge diagnostische waarde bij het syndroom van Alport. KS Chugh et al. (1993) vonden in een oogheelkundig onderzoek bij patiënten met het syndroom van Alport een afname van de gezichtsscherpte in 66,7% van de gevallen, anterieure lenticonus in 37,8%, netvliesvlekken in 22,2%, cataract in 20% en keratoconus in 6,7%.
Bij sommige kinderen met erfelijke nefritis, met name wanneer nierfalen ontstaat, wordt een aanzienlijke ontwikkelingsachterstand opgemerkt. Naarmate het nierfalen vordert, ontwikkelt zich arteriële hypertensie. Bij kinderen wordt dit vaker vastgesteld in de adolescentie en op oudere leeftijd.
Patiënten met erfelijke nefritis worden gekenmerkt door de aanwezigheid van diverse (meer dan 5-7) stigma's van bindweefseldysmorfogenese. De meest voorkomende bindweefselstigma's bij patiënten zijn hypertelorisme van de ogen, een hoog gehemelte, beetafwijkingen, een afwijkende vorm van de oorschelpen, een kromming van de pink aan de handen en een "sandaalspleet" aan de voeten. Erfelijke nefritis wordt gekenmerkt door een uniforme spreiding van dysmorfogenesestigma's binnen een familie, evenals een hoge frequentie van hun verspreiding onder verwanten van probands langs wiens lijn de ziekte wordt overgedragen.
In de vroege stadia van de ziekte wordt een geïsoleerde afname van de partiële nierfunctie waargenomen: transport van aminozuren, elektrolyten, concentratiefunctie, acidogenese; latere veranderingen beïnvloeden de functionele toestand van zowel het proximale als het distale deel van het nefron en worden gekenmerkt door gecombineerde partiële aandoeningen. Een afname van de glomerulaire filtratie treedt later op, vaker in de adolescentie. Naarmate erfelijke nefritis vordert, ontwikkelt zich bloedarmoede.
Erfelijke nefritis wordt gekenmerkt door een gefaseerd beloop van de ziekte: eerst een latent stadium of verborgen klinische symptomen, gemanifesteerd door minimale veranderingen in het urinaire syndroom, vervolgens treedt een geleidelijke decompensatie van het proces op met een afname van de nierfunctie met manifeste klinische symptomen (intoxicatie, asthenie, ontwikkelingsachterstand, bloedarmoede). Klinische symptomen treden meestal op ongeacht de gelaagdheid van de ontstekingsreactie.
Erfelijke nefritis kan zich op verschillende leeftijden manifesteren, afhankelijk van de werking van het gen, dat tot een bepaald moment in een onderdrukte toestand verkeert.
Classificatie
Er zijn drie soorten erfelijke nefritis
- Optie I - manifesteert zich klinisch als nefritis met hematurie, gehoorverlies en oogschade. Het beloop van nefritis is progressief en leidt tot de ontwikkeling van chronisch nierfalen. De overerving is dominant en gekoppeld aan het X-chromosoom. Morfologisch gezien wordt een verstoring van de structuur van de basale membraan, met verdunning en splitsing ervan, vastgesteld.
- Optie II - manifesteert zich klinisch als nefritis met hematurie zonder gehoorverlies. Het beloop van nefritis is progressief en leidt tot de ontwikkeling van chronisch nierfalen. De overerving is dominant en gekoppeld aan het X-chromosoom. Morfologisch gezien wordt er een verdunning van de glomerulaire capillaire basaalmembraan (met name de laminadensa) vastgesteld.
- Optie III - goedaardige familiaire hematurie. Het beloop is gunstig, chronisch nierfalen ontwikkelt zich niet. De overerving is autosomaal dominant of autosomaal recessief. Bij de autosomaal recessieve overerving wordt bij vrouwen een ernstiger beloop van de ziekte waargenomen.
Diagnose van het syndroom van Alport
De volgende criteria worden voorgesteld:
- de aanwezigheid van ten minste twee patiënten met nefropathie in elke familie;
- hematurie als voornaamste symptoom van nefropathie bij de proefpersoon;
- de aanwezigheid van gehoorverlies bij ten minste één familielid;
- ontwikkeling van chronisch nierfalen bij één of meer familieleden.
Bij de diagnostiek van diverse erfelijke en aangeboren aandoeningen wordt veel aandacht besteed aan een alomvattende aanpak van het onderzoek en vooral aan de gegevens die zijn verkregen bij het samenstellen van de stamboom van het kind. De diagnose van het syndroom van Alport wordt als geldig beschouwd wanneer 3 van de 4 typische symptomen bij de patiënt worden vastgesteld: de aanwezigheid van hematurie en chronisch nierfalen in de familie, de aanwezigheid van neurosensorisch gehoorverlies, visuele pathologie bij de patiënt, detectie van tekenen van splitsing van het glomerulaire basaalmembraan met een verandering in de dikte en onregelmatige contouren tijdens elektronenmicroscopische kenmerken van de biopsie.
Het onderzoek van de patiënt moet klinische en genetische onderzoeksmethoden omvatten; gerichte studie van de ziektegeschiedenis; algemeen onderzoek van de patiënt rekening houdend met diagnostisch significante criteria. In de compensatiefase kan pathologie alleen worden opgespoord door te focussen op syndromen zoals de aanwezigheid van een erfelijke belasting, hypotensie, meerdere stigma's van dysembryogenese, veranderingen in het urinaire syndroom. In de decompensatiefase kunnen extrarenale symptomen optreden, zoals ernstige intoxicatie, asthenie, vertraagde fysieke ontwikkeling, bloedarmoede, die zich manifesteren en intensiveren met een geleidelijke afname van de nierfunctie. Bij de meeste patiënten met een afname van de nierfunctie wordt het volgende waargenomen: verminderde acido- en aminogenese; 50% van de patiënten merkt een significante afname van de secretoire functie van de nieren op; beperkte reeks fluctuaties in de optische dichtheid van urine; verstoring van het filtratieritme, en vervolgens een afname van de glomerulaire filtratie. Er is sprake van chronisch nierfalen als de patiënt gedurende 3 tot 6 maanden of langer een verhoogd ureumgehalte in het bloedserum heeft (meer dan 0,35 g/l) en een daling van de glomerulaire filtratie tot 25% van de norm.
Differentiële diagnostiek van erfelijke nefritis dient primair te worden uitgevoerd bij de hematurische vorm van verworven glomerulonefritis. Verworven glomerulonefritis begint meestal acuut, binnen 2-3 weken na een infectie, met extrarenale verschijnselen, waaronder hypertensie vanaf de eerste dagen (bij erfelijke nefritis daarentegen hypotensie), verminderde glomerulaire filtratie bij het begin van de ziekte en geen aantasting van de gedeeltelijke tubulaire functies, terwijl dit bij erfelijke nefritis wel het geval is. Verworven glomerulonefritis treedt op met meer uitgesproken hematurie en proteïnurie, met een verhoogde bezinkingssnelheid (ESR). Typische veranderingen in de glomerulaire basaalmembraan, kenmerkend voor erfelijke nefritis, zijn van diagnostische waarde.
Differentiële diagnostiek van dysmetabole nefropathie wordt uitgevoerd bij chronisch nierfalen, in de familie klinisch aangetoonde heterogene nierziekten en er kan een spectrum van nefropathie voorkomen, variërend van pyelonefritis tot urolithiasis. Kinderen hebben vaak klachten van buikpijn en periodieke pijn tijdens het urineren, in het urinesediment - oxalaten.
Bij het vermoeden van erfelijke nefritis dient de patiënt te worden doorverwezen naar een gespecialiseerde nefrologieafdeling om de diagnose te verduidelijken.
Wat moeten we onderzoeken?
Hoe te onderzoeken?
Welke tests zijn nodig?
Met wie kun je contact opnemen?
Behandeling van het syndroom van Alport
Het regime omvat beperkingen op zware fysieke inspanning en blootstelling aan frisse lucht. Het dieet is compleet, met voldoende complete eiwitten, vetten en koolhydraten, rekening houdend met de nierfunctie. Van groot belang is het opsporen en behandelen van chronische infectiehaarden. De volgende medicijnen worden gebruikt: ATP, cocarboxylase, pyridoxine (tot 50 mg/dag), carnitinechloride. De kuren worden 2-3 keer per jaar gegeven. Voor hematurie worden kruidengeneesmiddelen voorgeschreven - brandnetel, appelbessap, duizendblad.
Er zijn berichten in de buitenlandse en binnenlandse literatuur over behandeling met prednisolon en het gebruik van cytostatica. Het effect is echter moeilijk te beoordelen.
Bij chronisch nierfalen worden hemodialyse en niertransplantatie toegepast.
Er zijn geen specifieke (effectieve pathogenetische) therapiemethoden voor erfelijke nefritis. Alle behandelmaatregelen zijn gericht op het voorkomen en vertragen van de achteruitgang van de nierfunctie.
Het dieet moet evenwichtig en calorierijk zijn, rekening houdend met de nierfunctie. Bij afwezigheid van functionele stoornissen moet het dieet van het kind voldoende eiwitten, vetten en koolhydraten bevatten. Bij tekenen van nierfunctiestoornissen moet de hoeveelheid eiwitten, koolhydraten, calcium en fosfor beperkt worden, wat de ontwikkeling van chronisch nierfalen vertraagt.
Beperk de fysieke activiteit. Kinderen wordt afgeraden om te sporten.
Contact met besmettelijke patiënten moet worden vermeden om het risico op het ontwikkelen van acute luchtwegaandoeningen te verminderen. Sanering van chronische infectiehaarden is noodzakelijk. Preventieve vaccinaties worden niet uitgevoerd bij kinderen met erfelijke nefritis; vaccinatie is alleen mogelijk bij epidemiologische indicaties.
Hormonale en immunosuppressieve therapie bij erfelijke nefritis is niet effectief. Er zijn aanwijzingen voor enig positief effect (vermindering van proteïnurie en vertraging van de ziekteprogressie) bij langdurig, meerjarig gebruik van ciclosporine A en ACE-remmers.
Bij de behandeling van patiënten worden medicijnen gebruikt die de stofwisseling verbeteren:
- pyridoxine - 2-3 mg/kg/dag in 3 doses gedurende 4 weken;
- cocarboxylase - 50 mg intramusculair om de dag, in totaal 10-15 injecties;
- ATP - 1 ml intramusculair om de dag, 10-15 injecties;
- vitamine A - 1000 IE/jaar/dag in 1 dosis gedurende 2 weken;
- Vitamine E - 1 mg/kg/dag in 1 dosis gedurende 2 weken.
Dit type therapie verbetert de algemene toestand van de patiënt, vermindert tubulaire disfuncties en wordt in kuren van 3 keer per jaar uitgevoerd.
Levamisol kan worden gebruikt als immunomodulator: 2 mg/kg/dag 2-3 keer per week met pauzes van 3-4 dagen tussen de doses.
Onderzoek toont aan dat hyperbare oxygenatie een positief effect heeft op de ernst van hematurie en nierfunctiestoornissen.
De meest effectieve methode voor de behandeling van erfelijke nefritis is tijdige niertransplantatie. In dit geval treedt er geen recidief van de ziekte op bij de transplantatie; in een klein percentage van de gevallen (ongeveer 5%) kan er nefritis ontstaan in de getransplanteerde nier, geassocieerd met antigenen tegen de glomerulaire basaalmembraan.
Een veelbelovende richting is prenatale diagnostiek en genetische manipulatie. Dierexperimenten tonen een hoge efficiëntie aan van het overbrengen van normale genen die verantwoordelijk zijn voor de synthese van type IV collageen-alfaketens naar nierweefsel, waarna de synthese van normale collageenstructuren wordt waargenomen.
Voorspelling
De prognose bij erfelijke nefritis is altijd ernstig.
Prognostisch ongunstige criteria voor het beloop van erfelijke nefritis zijn:
- mannelijk geslacht;
- vroege ontwikkeling van chronisch nierfalen bij familieleden;
- proteïnurie (meer dan 1 g/dag);
- verdikking van de glomerulaire basaalmembranen volgens microscopie;
- akoestische neuritis;
- deletie in het Col4A5-gen.
De prognose voor goedaardige familiaire hematurie is gunstiger.
Использованная литература