Medisch expert van het artikel

Nieuwe publicaties
Ziekte van Charcot-Marie-Tooth.
Laatst beoordeeld: 12.07.2025

Alle iLive-inhoud wordt medisch beoordeeld of gecontroleerd op feiten om zo veel mogelijk feitelijke nauwkeurigheid te waarborgen.
We hebben strikte richtlijnen voor sourcing en koppelen alleen aan gerenommeerde mediasites, academische onderzoeksinstellingen en, waar mogelijk, medisch getoetste onderzoeken. Merk op dat de nummers tussen haakjes ([1], [2], etc.) klikbare links naar deze studies zijn.
Als u van mening bent dat onze inhoud onjuist, verouderd of anderszins twijfelachtig is, selecteert u deze en drukt u op Ctrl + Enter.
Peroneale musculaire atrofie, syndroom van Charcot-Marie-Tooth of ziekte van Charcot-Marie-Tooth, is een groep chronische erfelijke ziekten waarbij de perifere zenuwen beschadigd raken.
Volgens de ICD-10, in de sectie over ziekten van het zenuwstelsel, is de code voor deze ziekte G60.0 (erfelijke motorische en sensorische neuropathie). De ziekte staat ook op de lijst van weesziekten.
Epidemiologie
Volgens klinische statistieken bedraagt de prevalentie van alle typen van de ziekte van Charcot-Marie-Tooth 19 gevallen per 100.000 inwoners (volgens andere bronnen één geval per 2,5-10.000 inwoners).
CMT type 1 is verantwoordelijk voor ongeveer twee derde van de gevallen (één geval per 5.000 tot 7.000 inwoners), en bijna 70% daarvan wordt geassocieerd met duplicatie van het PMP22-gen. Wereldwijd lijden meer dan 1,2 miljoen mensen aan dit type ziekte.
De incidentie van CMT type 4 wordt geschat op 1-5 gevallen per 10.000 kinderen. [ 1 ]
Oorzaken Ziekte van Charcot-Marie-Tooth
Volgens de classificatie van polyneuropathische syndromen verwijst peroneale (fibulaire) musculaire atrofie, Charcot-Marie-Tooth neurale amyotrofie of Charcot-Marie-Tooth-ziekte (afgekort als CMT) naar genetisch bepaalde motorisch-sensorische polyneuropathieën. [ 2 ]
Dat wil zeggen dat de oorzaken van het optreden ervan genetische mutaties zijn. Afhankelijk van de aard van de genetische afwijkingen worden de belangrijkste typen of vormen van dit syndroom onderscheiden: demyeliniserend en axonaal. De eerste groep omvat de ziekte van Charcot-Marie-Tooth type 1 (CMT1), die ontstaat door duplicatie van het PMP22-gen op chromosoom 17, dat codeert voor transmembraan perifeer myeline-eiwit 22. Als gevolg hiervan treedt segmentale demyelinisatie van de axonschede (uitlopers van zenuwcellen) op en neemt de snelheid van de zenuwsignaalgeleiding af. Daarnaast kunnen er mutaties in enkele andere genen voorkomen.
De axonale vorm is de ziekte van Charcot-Marie-Tooth type 2 (CMT2), die de axonen zelf aantast en gepaard gaat met pathologische veranderingen in het MFN2-gen op locus 1p36.22. Dit gen codeert voor het membraaneiwit mitofusine-2, dat nodig is voor de fusie van mitochondriën en de vorming van functionele mitochondriale netwerken in perifere zenuwcellen. Er bestaan meer dan een dozijn subtypes van CMT2 (met mutaties in specifieke genen).
Opgemerkt moet worden dat er momenteel meer dan honderd genen zijn geïdentificeerd waarvan de erfelijke schade verschillende subtypen van de ziekte van Charcot-Marie-Tooth veroorzaakt. Mutaties in het RAB7-gen leiden bijvoorbeeld tot CMT type 2B; een verandering in het SH3TC2-gen (dat codeert voor een van de membraaneiwitten van Schwann-cellen) veroorzaakt CMT type 4C, dat zich in de kindertijd manifesteert en wordt gekenmerkt door demyelinisatie van motorische en sensorische neuronen (er zijn twaalf en een half vormen van type 4 van deze ziekte).
Een zeldzaam type 3 CMT (ook wel Dejerine-Sottas-syndroom genoemd) begint zich te ontwikkelen in de vroege kinderjaren en wordt veroorzaakt door mutaties in de genen PMP22, MPZ, EGR2 en andere genen.
Wanneer CMT type 5 op de leeftijd van 5-12 jaar optreedt, wordt niet alleen motorische neuropathie (in de vorm van spastische paraparese van de onderste ledematen) waargenomen, maar ook schade aan de oogzenuw en de gehoorzenuw.
Spierzwakte en optische atrofie (met verlies van gezichtsvermogen) en evenwichtsproblemen zijn kenmerkend voor CMT type 6. Bij de ziekte van Charcot-Marie-Tooth type 7 is er niet alleen sprake van motorisch-sensorische neuropathie, maar ook van een netvliesziekte in de vorm van retinitis pigmentosa.
De X-gebonden CMT of de ziekte van Charcot-Marie-Tooth met tetraparese van de ledematen (zwakte in de beweging van zowel de armen als de benen) komt vaker voor bij mannen. Het is een demyeliniserend type en wordt vermoedelijk veroorzaakt door een mutatie in het GJB1-gen op de lange arm van het X-chromosoom, dat codeert voor connexine 32, een transmembraaneiwit van Schwann-cellen en oligodendrocyten dat de transmissie van zenuwsignalen reguleert. [ 3 ]
Risicofactoren
De belangrijkste risicofactor voor CMT is de familiegeschiedenis van de ziekte, dat wil zeggen bij naaste verwanten.
Volgens genetici is de kans op een kind dat deze ziekte ontwikkelt 25% als beide ouders drager zijn van het autosomaal recessieve gen voor de ziekte van Charcot-Marie-Tooth. De kans dat het kind drager is van dit gen (maar geen symptomen vertoont) wordt geschat op 50%.
Bij X-gebonden overerving (wanneer het gemuteerde gen zich op het X-chromosoom van de vrouw bevindt) is er een kans van 50% dat de moeder het gen doorgeeft aan haar zoon, die CMT zal ontwikkelen. De ziekte treedt mogelijk niet op bij de geboorte van een meisje, maar de zonen (kleinkinderen) van de dochter kunnen het defecte gen erven, waardoor de ziekte zich wel zal ontwikkelen.
Pathogenese
Bij elke vorm van de ziekte van Charcot-Marie-Tooth wordt de pathogenese veroorzaakt door een erfelijke afwijking van de perifere zenuwen: motorische (bewegings-) en sensorische (gevoeligheids-) zenuwen.
Bij het CMT-type is er sprake van demyeliniserende mutaties, wat betekent dat de vernietiging of het defect van de myelineschede die de axonen van de perifere zenuwen beschermt, leidt tot een vertraging van de overdracht van zenuwimpulsen in het perifere zenuwstelsel - tussen de hersenen, spieren en zintuigen.
Bij de axonale vorm van de ziekte zijn de axonen zelf aangetast, waardoor de zenuwsignalen niet meer sterk genoeg zijn om de spieren en zintuigen volledig te stimuleren.
Lees ook:
Hoe wordt het Charcot-Marie-Tooth-syndroom overgedragen? De defecte genen kunnen autosomaal dominant of autosomaal recessief worden overgeërfd.
Het meest voorkomende type, autosomaal dominante overerving, treedt op wanneer er één kopie van het gemuteerde gen aanwezig is (gedragen door één van de ouders). De kans dat CMT aan elk geboren kind wordt doorgegeven, wordt geschat op 50%. [ 4 ]
Bij autosomaal recessieve overerving zijn er twee kopieën van het defecte gen nodig (één van elke ouder die geen symptomen van de ziekte vertoont) om de ziekte te ontwikkelen.
In 40-50% van de gevallen treedt autosomaal dominante erfelijke demyelinisatie op, d.w.z. CMT type 1; in 12-26% van de gevallen axonale CMT, d.w.z. type 2. En in 10-15% van de gevallen wordt X-gebonden overerving waargenomen. [ 5 ]
Symptomen Ziekte van Charcot-Marie-Tooth
De eerste tekenen van deze ziekte openbaren zich doorgaans in de kindertijd en adolescentie en ontwikkelen zich geleidelijk gedurende het hele leven, hoewel het syndroom zich later kan openbaren. De combinatie van symptomen is variabel en de snelheid waarmee de ziekte zich ontwikkelt, evenals de ernst ervan, is onmogelijk te voorspellen.
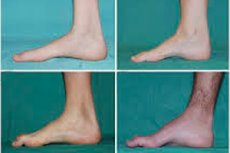
Typische symptomen in het beginstadium zijn onder meer toegenomen algemene vermoeidheid; verminderde tonus (zwakte) van de spieren in de voeten, enkels en scheenbenen; gebrek aan reflexen. Dit bemoeilijkt de voetbeweging en leidt tot dysbasie (loopstoornis) in de vorm van een hogere beenopstand, vaak met frequent struikelen en vallen. Tekenen van de ziekte van Charcot-Marie-Tooth bij een jong kind kunnen zijn: uitgesproken onhandigheid en leeftijdsgebonden loopproblemen die gepaard gaan met een bilaterale klapvoet. Voetmisvormingen zijn ook kenmerkend: een hoge wreef (holle voet) of ernstige platvoeten, en kromme (hamervormige) tenen.
Bij het op de tenen lopen tegen een achtergrond van spierhypotonie kan de neuroloog vermoeden dat het kind CMT type 4 heeft, een stoornis waarbij kinderen tegen de tijd dat ze in de adolescentie zijn, niet meer kunnen lopen.
Naarmate de ziekte vordert, breiden spieratrofie en spierzwakte zich uit naar de bovenste ledematen, waardoor fijne motoriek en normale handbewegingen moeilijk worden. Verminderde tastzin en het vermogen om warmte en kou te voelen, evenals gevoelloosheid in de voeten en handen, wijzen op schade aan de axonen van de sensorische zenuwen.
Bij de ziekte van Charcot-Marie-Tooth type 3 en 6, die zich in de kindertijd manifesteert, worden sensorische ataxie (verminderde coördinatie van bewegingen en evenwicht), spiertrekkingen en tremoren, schade aan de aangezichtszenuw, oogzenuwatrofie met nystagmus en gehoorverlies waargenomen.
In een later stadium kan er sprake zijn van oncontroleerbaar trillen (tremor) en vaak ook spierkrampen. Problemen met bewegen kunnen leiden tot pijn: spier-, gewrichts- en neuropathische pijn.
Complicaties en gevolgen
De ziekte van Charcot-Marie-Tooth kan complicaties en gevolgen hebben, zoals:
- vaker verstuikingen en breuken;
- contracturen geassocieerd met verkorting van periarticulaire spieren en pezen;
- scoliose (kromming van de wervelkolom);
- ademhalingsproblemen – wanneer de zenuwvezels die de spieren van het middenrif innerveren beschadigd zijn:
- verlies van het vermogen om zelfstandig te bewegen.
Diagnostics Ziekte van Charcot-Marie-Tooth
De diagnose bestaat uit klinisch onderzoek, anamnese (inclusief familiegeschiedenis), neurologisch en systemisch onderzoek.
Er worden tests uitgevoerd om het bewegingsbereik, de gevoeligheid en de peesreflexen te controleren. De zenuwgeleiding kan worden beoordeeld met behulp van instrumentele diagnostiek – elektromyografie of elektroneuromyografie. Echografie of MRI kan ook nodig zijn. [ 6 ]
Genetische of DNA-testen om de meest voorkomende genetische mutaties die CMT veroorzaken in een bloedmonster op te sporen, zijn beperkt omdat DNA-testen momenteel niet voor alle typen van de ziekte beschikbaar zijn. Zie Genetische tests voor meer informatie.
In sommige gevallen wordt een biopsie van de perifere zenuw (meestal de nervus suralis) uitgevoerd.
Differentiële diagnose
De differentiële diagnose omvat andere perifere neuropathieën, de spierdystrofie van Duchenne, myelopathische en myasthenische syndromen, diabetische neuropathie, myelopathieën bij multiple sclerose en amyotrofische laterale sclerose, het syndroom van Guillain-Barré, trauma aan de peroneale zenuw en de atrofie ervan (ook wanneer deze bekneld raakt tussen de lumbale tussenwervelschijven), schade aan de kleine hersenen of de thalamus, evenals bijwerkingen van chemotherapie (tijdens behandeling met cytostatica zoals Vincristine of Paclitaxel). [ 7 ]
Met wie kun je contact opnemen?
Behandeling Ziekte van Charcot-Marie-Tooth
Tegenwoordig bestaat de behandeling van deze erfelijke ziekte uit oefentherapie (gericht op het versterken en rekken van de spieren); ergotherapie (die patiënten met spierzwakte in de handen helpt); en het gebruik van orthopedische hulpmiddelen om het lopen te vergemakkelijken. Indien nodig worden pijnstillers of anti-epileptica voorgeschreven. [ 8 ]
Bij ernstige platvoeten kan een osteotomie worden uitgevoerd, en bij hieldeformatie is een chirurgische correctie geïndiceerd – artrodese. [ 9 ]
Er wordt onderzoek gedaan naar zowel de genetische component van de ziekte als de behandelmethoden. Het gebruik van stamcellen, bepaalde hormonen, lecithine of ascorbinezuur heeft nog geen positieve resultaten opgeleverd.
Maar dankzij recent onderzoek zou er in de nabije toekomst wel eens iets nieuws kunnen verschijnen in de behandeling van de ziekte van Charcot-Marie-Tooth. Zo ontwikkelt het Franse bedrijf Pharnext sinds 2014, en sinds medio 2019 lopen er klinische studies naar het medicijn PXT3003 voor de behandeling van CMT type 1 bij volwassenen. PXT3003 onderdrukt de verhoogde expressie van het PMP22-gen, verbetert de myelinisatie van perifere zenuwen en verlicht neuromusculaire symptomen.
Specialisten van het medische bedrijf Sarepta Therapeutics (VS) werken aan een gentherapie voor de ziekte van Charcot-Marie-Tooth type 1. Deze therapie maakt gebruik van een onschadelijk adeno-geassocieerd virus (AAV) van het geslacht Dependovirus met een lineair enkelstrengs DNA-genoom. Dit gen zal het NTF3-gen in het lichaam brengen. Dit gen codeert voor het neurotrofine-3 (NT-3)-eiwit dat nodig is voor het functioneren van Schwann-zenuwcellen.
Helixmith zal eind 2020 beginnen met klinische proeven met de in Zuid-Korea ontwikkelde gentherapie Engensis (VM202) voor de behandeling van spiersymptomen bij CMT type 1. [ 10 ]
Het voorkomen
Preventie van CMT kan bestaan uit genetische counseling van aanstaande ouders, vooral als iemand in het paar een familiegeschiedenis van de ziekte heeft. Er zijn echter gevallen van de novo puntgenmutaties vastgesteld, dat wil zeggen, zonder dat de ziekte in de familiegeschiedenis voorkomt.
Tijdens de zwangerschap kan de kans op de ziekte van Charcot-Marie-Tooth bij het toekomstige kind worden gecontroleerd door middel van een vlokkentest (vanaf 10 tot 13 weken zwangerschap) en een analyse van het vruchtwater (vanaf 15 tot 18 weken).
Prognose
Over het algemeen hangt de prognose voor verschillende vormen van de ziekte van Charcot-Marie-Tooth af van de klinische ernst, maar in alle gevallen verloopt de ziekte langzaam. Veel patiënten hebben een handicap, hoewel dit de levensverwachting niet verkort.