Medisch expert van het artikel

Nieuwe publicaties
Neuroblastoom bij kinderen: oorzaken, diagnose, behandeling
Laatst beoordeeld: 04.07.2025

Alle iLive-inhoud wordt medisch beoordeeld of gecontroleerd op feiten om zo veel mogelijk feitelijke nauwkeurigheid te waarborgen.
We hebben strikte richtlijnen voor sourcing en koppelen alleen aan gerenommeerde mediasites, academische onderzoeksinstellingen en, waar mogelijk, medisch getoetste onderzoeken. Merk op dat de nummers tussen haakjes ([1], [2], etc.) klikbare links naar deze studies zijn.
Als u van mening bent dat onze inhoud onjuist, verouderd of anderszins twijfelachtig is, selecteert u deze en drukt u op Ctrl + Enter.
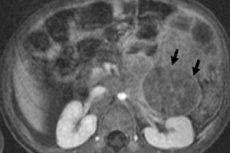
Eén van de meest voorkomende extracraniële neoplasmata in de kinderoncologie is het neuroblastoom bij kinderen. Dit is een embryonale, kwaadaardige tumor van de neuroblasten van de neurale lijst, dat wil zeggen van embryonale (onvolwassen) zenuwcellen van het sympathische zenuwstelsel.
Epidemiologie
Volgens statistieken van de International Neuroblastoma Risk Group (INRG) is neuroblastoom verantwoordelijk voor ongeveer 8% van alle oncologische ziekten bij kinderen wereldwijd en staat het op de derde plaats in prevalentie, na leukemie en hersentumoren.
Volgens andere gegevens is neuroblastoom verantwoordelijk voor ongeveer 28% van alle vormen van kanker bij zuigelingen. Meer dan een derde van de gevallen van neuroblastoom wordt vastgesteld bij kinderen jonger dan één jaar; de gemiddelde leeftijd bij diagnose is 19-22 maanden. Meer dan 90% van de gediagnosticeerde gevallen komt voor bij kinderen van twee tot vijf jaar (met een overwicht aan jongens); de piekincidentie wordt waargenomen op de leeftijd van twee tot drie jaar, en minder dan 10% van de gevallen bij kinderen ouder dan vijf jaar.
Oorzaken neuroblastomen
Bij het bestuderen van de oorzaken van neuroblastoom zijn onderzoekers tot de conclusie gekomen dat deze tumor bij kinderen ontstaat door sporadische genetische mutaties tijdens de embryogenese of vroege postnatale ontwikkeling. De oorzaak van deze genveranderingen is echter onbekend, aangezien er geen invloed van teratogene omgevingsfactoren is vastgesteld.
Deze tumoren kunnen overal voorkomen, bijvoorbeeld in het mediastinum, de nek, de buik, de bijnieren, de nieren, de wervelkolom en het bekken.
In zeldzame gevallen kan neuroblastoom bij zuigelingen gepaard gaan met een erfelijke mutatie. Met name een mutatie in het gen van het membraaneiwit CD246 op chromosoom 2 – het enzym tyrosinekinase ALK, dat zorgt voor intercellulaire communicatie en een belangrijke rol speelt in de werking van het zenuwstelsel; en in het gen van het eiwit PHOX2B (op chromosoom 4), dat betrokken is bij de rijping van zenuwcellen.
Neuroblastoom kan ook verband houden met kinderneurofibromatose type 1,het syndroom van Beckwith-Wiedemann en hyperinsulinemische hypoglykemie (nesidioblastose pancreatitis).
Risicofactoren
Tegenwoordig worden erfelijkheid en de aanwezigheid van deze tumor in de familiegeschiedenis erkend als risicofactoren voor de ontwikkeling van neuroblastoom bij kinderen, evenals aangeboren afwijkingen die verband houden met genmutaties tijdens de intra-uteriene ontwikkeling. Dit geldt met name voor gevallen waarin meerdere neoplasmata in verschillende organen ontstaan.
Onderzoekers hebben nog geen van de externe factoren geïdentificeerd die het risico op deze tumor vergroten.
Pathogenese
Het ontwikkelingsmechanisme van neuroblastomen wordt veroorzaakt door verstoringen in de differentiatie en rijping van neurale lijstcellen – bilaterale cellijnen die zich aan de randen van de neurale buis vormen vanuit de ectodermale kiemlaag van het menselijk embryo. Deze cellen migreren (verplaatsen) en differentiëren tot vele soorten cellen: sensorische en autonome neuronen, neuro-endocriene cellen en cellen van het bijniermerg, cellen van het craniofaciale kraakbeen en botten, evenals pigmentcellen.
Bij neuroblastoom rijpen de gemigreerde neuroblasten niet, maar blijven ze groeien en delen, waardoor een tumor ontstaat. De pathogenese van de vorming ervan wordt geassocieerd met de volgende genmutaties:
- met duplicatie van een deel van de chromosoomsequentie of duplicatie van segmenten van het LMO1-gen op chromosoom 11, dat codeert voor het RBTN1-eiwit in de neurale lijstcellen van het embryo;
- met een verandering in het aantal kopieën van het NBPF10-gen op chromosoom 1q21.1, dat codeert voor het eiwit DUF1220, dat de proliferatie van menselijke neurale stamcellen reguleert. Deze aandoeningen leiden tot een duplicatie van dit chromosoom of tot deletie ervan - de afwezigheid van een deel van het DNA;
- met veranderingen in het tumorsuppressorgen ATRX (op chromosoom Xq21.1);
- Met de aanwezigheid van extra kopieën (amplificatie) van het N-Myc transcriptiefactorgen op chromosoom 2, dat codeert voor een van de transcriptiefactoren (DNA-bindend eiwit) die de activiteit van andere genen reguleert en de proliferatie van voorlopercellen regelt tijdens de vorming van eiwitten voor de vorming van weefsels en organen van de foetus. Amplificatie van dit gen verandert het in een oncogen, wat leidt tot een verstoring van de celcyclus, verhoogde celproliferatie en tumorvorming.
Symptomen neuroblastomen
De eerste tekenen van neuroblastoom zijn niet-specifiek en kunnen bestaan uit verlies van eetlust (en gewichtsverlies), vermoeidheid bij het eten, koorts en gewrichtspijn.
De klinische symptomen zijn afhankelijk van de locatie van de primaire tumor en de aanwezigheid van uitzaaiingen (die in 60-73% van de gevallen voorkomen).
Zeer vaak is een primair neuroblastoom gelokaliseerd in het bijniermerg, dat een vergelijkbare oorsprong heeft als zenuwcellen. Bij kinderen jonger dan één jaar wordt in 35-40% van de gevallen een bijnierneuroblastoom vastgesteld. De symptomen zijn onder andere buikpijn, koorts, gewichtsverlies, botpijn, bloedarmoede of gelijktijdig het syndroom van Pepper: diffuse leverschade met ernstige hepatomegalie en respiratoir distresssyndroom.
Retroperitoneale neuroblastoom of retroperitoneale neuroblastoom bij kinderen begint tijdens de groei op de blaas of de darmen te drukken, waardoor problemen met urineren of ontlasting kunnen ontstaan, zwelling van de benen (bij jongens zwelt het scrotum op).
Neuroblastoom van het mediastinum bij kinderen (mediastinaal neuroblastoom) drukt vaak op de vena cava superior, wat zwelling van het gezicht, de nek, de armen en de bovenborst kan veroorzaken (de huid wordt blauwrood met onderhuidse knobbeltjes). Hoesten en piepen, ademhalingsproblemen (kortademigheid) of slikproblemen (dysfagie) komen voor; vergrote lymfeklieren worden opgemerkt in de nek, boven het sleutelbeen en in de oksels.
De verspreiding van tumorcellen naar het beenmerg leidt tot bloedarmoede, trombocytopenie en leukopenie met een neiging tot bloedingen.
En bij uitzaaiingen in het periorbitale gebied ontstaan donkere kringen of blauwe plekken rond de ogen. Een dergelijke tumor kan ook hoofdpijn en duizeligheid, exoftalmie (uitpuilende oogbollen) en, door compressie van de zenuwuiteinden, hangende oogleden (ptosis) en een verkleining van de pupillen (miosis) veroorzaken.
Abdominaal neuroblastoom of neuroblastoom van de buikholte bij kinderen leidt tot de vorming van voelbare afsluitingen in de buik, een opgezette buik, verlies van eetlust, constipatie en een verhoogde bloeddruk. Een tumor die op het ruggenmerg of de zenuwwortel drukt, kan leiden tot gevoelloosheid en zwakte in de ledematen, en tot het onvermogen om te staan, kruipen of lopen. Als botten zijn aangetast, kan botpijn optreden.
Bij een tumor in stadium 3-4 in de buikholte met schade aan de lymfeklieren kunnen tumorcellen het nierparenchym binnendringen. Vervolgens ontstaat bij kinderen een uitgebreid neuroblastoom van de nier, wat leidt tot verstoring van de nierfunctie.
Stages
- Neuroblastoom in stadium 1 is een primaire tumor die geïsoleerd en gelokaliseerd is in één gebied van het lichaam. De lymfeklieren aan beide kanten worden niet aangetast.
- Neuroblastoom stadium 2. In stadium 2A is de primaire tumor beperkt tot één gebied, maar groot; bilaterale lymfeklieren zijn niet aangetast. In stadium 2B zijn de lymfeklieren aan de lichaamszijde waar de tumor zich bevindt positief voor uitzaaiingen.
- Neuroblastoom stadium 3: de primaire tumor kruist het ruggenmerg of de middenlijn van het lichaam, er worden unilaterale of bilaterale metastasen gevonden in de lymfeklieren.
- Neuroblastoom stadium 4: de tumor is uitgezaaid naar lymfeklieren, beenmerg, botten, lever of andere organen. Stadium 4S wordt vastgesteld bij kinderen jonger dan één jaar met een gelokaliseerde primaire tumor, met uitzaaiingen naar de huid, lever of het beenmerg.
Internationaal Neuroblastoom Risicostadiëringssysteem (INRGSS)
Bij INRGSS wordt gebruikgemaakt van op beeldvorming gebaseerde risicofactoren (IDRF's). Dit zijn factoren die bij beeldvormend onderzoek zichtbaar zijn en die kunnen betekenen dat een tumor moeilijker te verwijderen is.
INRGSS verdeelt neuroblastomen in 4 stadia:
- L1: De tumor heeft zich niet verspreid vanaf de start en is niet uitgegroeid tot vitale structuren. De tumor is beperkt tot één deel van het lichaam, zoals de nek, borst of buik.
- L2: De tumor heeft zich niet ver van de oorsprong verspreid (gemetastaseerd) (bijvoorbeeld van de linkerkant van de buik naar de linkerkant van de borstkas), maar heeft wel minstens één IDRF.
- M: De tumor is uitgezaaid naar een verafgelegen deel van het lichaam (met uitzondering van tumoren in het MS-stadium).
- MS: Metastatische ziekte bij kinderen jonger dan 18 maanden, waarbij de kanker zich alleen heeft verspreid naar de huid, de lever en/of het beenmerg.
Complicaties en gevolgen
Neuroblastoom wordt gekenmerkt door complicaties en gevolgen zoals:
- uitzaaiing (metastase) naar de lymfeklieren, het beenmerg, de lever, de huid en de botten;
- compressie van het ruggenmerg (wat pijn kan veroorzaken en tot verlamming kan leiden);
- ontwikkeling van paraneoplastisch syndroom (als gevolg van de werking van bepaalde chemicaliën die door de tumor worden afgescheiden, evenals het antigeen disialoganglioside GD2 dat door de cellen ervan tot expressie wordt gebracht), wat zich manifesteert door snelle onwillekeurige oogbewegingen, verminderde coördinatie, spierkrampen en diarree;
- recidieven na voltooiing van de primaire therapie (zoals de klinische praktijk laat zien, hebben hoog-risico neuroblastomen in 50% van de gevallen een recidief).
Diagnostics neuroblastomen
Om bij een kind het vermoeden van neuroblastoom te diagnosticeren, zijn onderzoek, laboratoriumtests en beeldvorming nodig.
Er worden bloed- en urineonderzoeken gedaan naar catecholamines (noradrenaline en dopamine) en homovanilline- of vanillylamandelzuur (gevormd tijdens de stofwisseling van deze hormonen); er wordt een bloedtest afgenomen voor neurospecifieke enolase, een enzyme-linked immunosorbent assay (ELISA) van het bloedserum en een beenmergonderzoek (waarvan een monster wordt afgenomen door middel van aspiratie). Er wordt een DNA-test uitgevoerd om mutaties vast te stellen en er wordt een biopsie uitgevoerd voor cytomorfologisch onderzoek van het tumorweefsel.
Nadat de biopten zijn afgenomen, worden ze naar een laboratorium gestuurd waar ze onder een microscoop worden bekeken door een patholoog (een arts die speciaal is opgeleid in het identificeren van kankercellen). Vaak worden er ook speciale laboratoriumtests op de monsters uitgevoerd om aan te tonen of de tumor neuroblastoom is.
Als het om een neuroblastoom gaat, kunnen laboratoriumtests ook helpen bepalen hoe snel de tumor groeit of zich verspreidt, en welke behandelingen het beste werken.
Instrumentele diagnostiek visualiseert het neoplasma met behulp van echografie, röntgenfoto's, MRI of CT, PET met de introductie van 18F-fluorodeoxyglucose- of MIBG-scanning - scintigrafie met metaiodobenzylguanidine. [ 1 ]
Differentiële diagnose
De differentiële diagnose omvat goedaardig ganglioneuroom, ganglioneuroblastoom, rhabdomyosarcoom en nefroblastoom.
Met wie kun je contact opnemen?
Behandeling neuroblastomen
Bij neuroblastoom hangt de behandeling af van de risicogroep van de patiënt (stadium van het tumorproces), de lokalisatie van de tumor, de genomische kenmerken van de tumorcellen en de leeftijd van het kind. De behandeling kan bestaan uit monitoring, chirurgie, chemotherapie, radiotherapie, immunotherapie en hematopoëtische stamceltransplantatie.
Neoadjuvante of adjuvante (pre- of postoperatieve) chemotherapie voor neuroblastoom bij kinderen wordt, net als elke chemotherapie voor kanker, in kuren gegeven: het medicijn wordt meerdere dagen achter elkaar toegediend, gevolgd door een rustpauze zodat het lichaam kan herstellen. De kuren worden meestal om de drie tot vier weken herhaald.
De volgende medicijnen (en hun combinaties) worden gebruikt: Cyclofosfamide, Cisplatine of Carboplatine, Doxorubicine (Adriamycine), Vincristine, Etoposide.
Vaak voorkomende bijwerkingen van chemotherapie zijn haaruitval, verlies van eetlust, vermoeidheid, misselijkheid en braken, aften, diarree of constipatie. Chemotherapie kan het beenmerg beschadigen en het aantal bloedcellen verlagen.
Gerichte immunotherapie (gericht op het tumorantigeen GD2) maakt gebruik van geneesmiddelen uit de groep monoklonale antilichamen (anti-GD2 MAb) dinutuximab (unituxin) en naxitamab. Deze worden intraveneus toegediend via een verlengde infusie, in combinatie met granulocyt-macrofaag koloniestimulerende factor (cytokine GM-CSF) en interleukine-2.
Bijwerkingen van deze medicijnen zijn onder meer pijn (vaak zeer hevig), een lage bloeddruk, een verhoogde hartslag, kortademigheid (met mogelijke zwelling van de luchtwegen), een verhoogde temperatuur, misselijkheid, braken en diarree, en veranderingen in de cellulaire en minerale samenstelling van het bloed.
Om het risico op terugkeer van kanker na chemotherapie met hoge doseringen en stamceltransplantatie te verminderen, worden kinderen met een hoog-risico neuroblastoom behandeld met systemische retinoïden, 13-cis-retinoïnezuur (isotretinoïne). [ 2 ]
Chirurgische behandeling van neuroblastoom – verwijdering van de tumor, bijvoorbeeld open adrenalectomie of laparoscopische resectie van adrenaal neuroblastoom; lymfectomie (verwijdering van de aangetaste lymfeklieren), enz. [ 3 ]
Bij neuroblastoom met een hoog risico kan radiotherapie worden gebruikt.[ 4 ]
Het voorkomen
Gezien de oorzaken van neuroblastoom bij kinderen is genetische counseling bij het plannen van een zwangerschap wellicht de enige preventieve maatregel. Houd er echter rekening mee dat deze tumor slechts in 1-2% van de gevallen gepaard gaat met erfelijke mutaties.
Prognose
Bij infantiel neuroblastoom is er sprake van spontane regressie.
Prognostische markers
- Tumoren met een hoog risico, evenals neuroblastoom bij kinderen van alle leeftijden en alle stadia (behalve stadium 4S) – met verhoogde expressie van het N-MYC-gen en amplificatie van het N-Myc-oncogen – hebben een ongunstige prognose die de levensverwachting beïnvloedt.
- Tumorcellen die bepaalde delen van chromosoom 1 of 11 missen (ook wel 1p- of 11q-deleties genoemd) hebben een slechtere prognose. Een extra deel van chromosoom 17 (17q-winst) wordt ook geassocieerd met een slechtere prognose.
- Neuroblastoomcellen met een grote hoeveelheid DNA hebben een betere prognose, vooral bij kinderen jonger dan 2 jaar.
- Neuroblastomen met meer neurotrofine-receptoren, vooral de zenuwgroeifactorreceptor TrkA, hebben een betere prognose.
Overleving per risicogroep van de Childhood Oncology Group (COG)
- Laagrisicogroep: Kinderen in de laagrisicogroep hebben een 5-jaarsoverlevingskans van meer dan 95%.
- Intermediaire risicogroep: Kinderen in de intermediaire risicogroep hebben een 5-jaarsoverlevingskans van 90% tot 95%.
- Hoogrisicogroep: Kinderen in de hoogrisicogroep hebben na 5 jaar een overlevingskans van ongeveer 50%.
Ongeveer 15% van de sterfgevallen door kinderkanker wordt veroorzaakt door neuroblastoom. De overlevingskans op lange termijn voor deze hoogrisico-maligniteit bedraagt maximaal 40%. De totale vijfjaarsoverleving bedraagt 67-74%, 43% in de leeftijdsgroep van één tot vier jaar en meer dan 80% voor neuroblastoom dat in het eerste levensjaar wordt gediagnosticeerd.
Использованная литература