Medisch expert van het artikel

Nieuwe publicaties
Cystic fibrosis
Laatst beoordeeld: 23.04.2024

Alle iLive-inhoud wordt medisch beoordeeld of gecontroleerd op feiten om zo veel mogelijk feitelijke nauwkeurigheid te waarborgen.
We hebben strikte richtlijnen voor sourcing en koppelen alleen aan gerenommeerde mediasites, academische onderzoeksinstellingen en, waar mogelijk, medisch getoetste onderzoeken. Merk op dat de nummers tussen haakjes ([1], [2], etc.) klikbare links naar deze studies zijn.
Als u van mening bent dat onze inhoud onjuist, verouderd of anderszins twijfelachtig is, selecteert u deze en drukt u op Ctrl + Enter.
Cystic fibrosis - een erfelijke autosomaal recessieve monogene ziekte die wordt gekenmerkt door een verminderde secretie van exocriene klieren vitale organen met laesies in de eerste plaats van de luchtwegen en het spijsverteringsstelsel, ernstige en ongunstige prognose.
 [1]
[1]
Epidemiologie
De incidentie van cystische fibrose varieert van 1: 2.500 tot 1: 4.600 pasgeborenen. Jaarlijks worden in de wereld ongeveer 45000 patiënten met cystic fibrosis geboren. De frequentie van gen-dragers van cystic fibrosis 3-4%, over de hele wereld, zijn er ongeveer 275 miljoen mensen - .. De dragers van het gen, waarvan ongeveer 5 miljoen mensen het leven in Rusland, ongeveer 12,5 miljoen -. In CH landen.
Oorzaken cystische fibrose
Cystic fibrosis wordt overgedragen door een autosomaal recessief type. Het gen van cystische fibrose bevindt zich in het 7 autosoom, bevat 27 exons en bestaat uit 250.000 paren nucleotiden.
In één gen zijn vele mutaties mogelijk, die elk kenmerkend zijn voor een bepaalde populatie of een bepaalde geografische regio. Meer dan 520 mutaties worden beschreven, de meest voorkomende daarvan is delta-P-508, d.w.z. Vervanging van het aminozuur fenylalanine in 508 posities.
Pathogenese
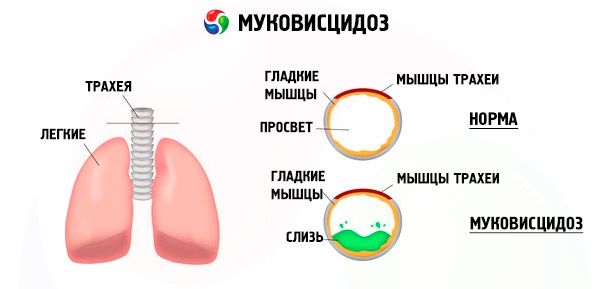
Als gevolg van mutaties van het cystic fibrosis-gen zijn de structuur en functie van het eiwit dat de CFTR-cystische fibrose transmembraanregulator wordt genoemd, verstoord. Dit eiwit speelt een rol van het chloridekanaal betrokken in water elektrolyt metabolisme epitheelcellen bronchopulmonaire, het maagdarmkanaal, pancreas, lever, voortplantingssysteem. Als gevolg van de schending van de functie en structuur van het CFTR-eiwit hopen chloorionen zich op in de cel. Cl - ). Dit leidt tot een verandering van de elektrische potentiaal in het lumen van de uitlaatkanalen, in een grote hoeveelheid natriumionen (Na daardoor invoeren + ) uit het lumen in de celstroom en verder versterkt de absorptie van water uit de pericellulaire ruimte.
Als gevolg van deze veranderingen wordt het geheim van de meeste klieren van externe secretie dikker, de evacuatie ervan wordt verstoord, wat leidt tot uitgesproken secundaire stoornissen in organen en systemen die het meest uitgesproken zijn in de bronchopulmonale en spijsverteringsstelsels.
De bronchiën ontwikkelen een chronische ontstekingsproces van variërende intensiteit sterk verstoorde functie van de trilhaarepitheel, slijm zeer viskeus, dik, moeilijk te evacueren wordt waargenomen stagnatie gevormd bronhiolo- en bronchiëctasie, die steeds vaker gebruikt in de tijd. Deze veranderingen leiden tot een toename van hypoxie en de vorming van een chronisch longhart.
Patiënten met cystische fibrose zijn extreem gepredisponeerd voor de ontwikkeling van een chronisch ontstekingsproces in het bronchopulmonale systeem. Dit is het gevolg van uitgesproken stoornissen in het systeem van lokale bronchopulmonale bescherming (verlaging van het niveau van IgA, interferon, fagocytische functie van alveolaire macrofagen en leukocyten).
Een belangrijke rol bij de ontwikkeling van chronische ontstekingen in het bronchopulmonale systeem is van alveolaire macrofagen. Ze produceren een groot aantal IL-8, wat de chemotaxis van neutrofielen in de bronchiale boom drastisch verhoogt. Neutrofielen hopen zich in grote hoeveelheden op in de bronchiën en leveren samen met epitheelcellen veel ontstekingsremmende cytokinen af, waaronder IL-1, 8, 6, tumornecrosefactor en leukotriënen.
Een belangrijke rol in de pathogenese van de laesie van het bronchopulmonale systeem wordt ook gespeeld door de hoge activiteit van het enzym elastase. Er zijn exogene en endogene elastase. De eerste wordt geproduceerd door de bacteriële flora (vooral de Pseudomonas aeruginosa), de tweede door neutrofiele leukocyten. Elastase vernietigt het epitheel en andere structurele elementen van de bronchiën, wat bijdraagt aan de verdere verstoring van het mucociliaire transport en de snelste vorming van bronchiectasieën.
Neutrofiele leukocyten scheiden ook andere proteolytische enzymen uit. Ze neutraliseren het effect van proteolytische enzymen en beschermen bijgevolg het bronchopulmonaire systeem tegen hun schadelijke effect van al-anpripinsine en de secretorische remmer van leukoproteasen. Helaas worden deze beschermende factoren bij patiënten met cystische fibrose echter onderdrukt door een aanzienlijke hoeveelheid neutrofielprotease.
Al deze omstandigheden dragen bij aan de introductie van een infectie in het bronchopulmonale systeem, de ontwikkeling van chronische purulente bronchitis. Voorts zij opgemerkt dat het defecte proteïne gecodeerd door het gen voor cystische fibrose, verandert de bedrijfstoestand van het bronchiale epitheel, waarbij de hechting van bacteriën begunstigt het bronchiale epitheel, met name Pseudomonas aeruginosa.
Samen met de pathologie van het bronchopulmonale systeem bij cystische fibrose, is er ook een duidelijke nederlaag van de pancreas, maag, dikke en dunne darm en lever.
Symptomen cystische fibrose
Cystic fibrosis manifesteert zich in een verscheidenheid van klinische symptomen. Bij pasgeborenen kan de ziekte zich manifesteren met een meconische ileus. Door het ontbreken of zelfs de volledige afwezigheid van trypsine, wordt het meconium zeer dicht, stroperig, hoopt zich op in het ileocecale gebied. Verdere ontwikkeling van ileus, die zich manifesteert met intense braken gal, winderigheid, door een gebrek aan meconium peritonitis symptomen, de snelle groei van de klinische verschijnselen van het syndroom van ernstige intoxicatie. Een kind kan in de eerste dagen van zijn leven sterven, tenzij er een dringende chirurgische ingreep wordt uitgevoerd.
In minder ernstige gevallen is een kenmerkend teken van cystische fibrose een overvloedige, frequente stoelgang, zalf, met veel vet, met een zeer onaangename geur. In 1/3 patiënten is er een prolaps van het rectum.
Vervolgens blijft intestinale disfunctie bestaan bij patiënten, malabsorptiesyndroom ontwikkelt zich, ernstige schending van lichamelijke ontwikkeling, ernstige hypovitaminose.
In het eerste of tweede levensjaar van het kind verschijnen symptomen van een bronchopulmonair systeem (milde vorm van de ziekte), wat zich uit in een hoest die extreem uitgesproken kan zijn en lijkt op een hoest in kinkhoest. Hoest gepaard met cyanose, kortademigheid, afscheiding van dik sputum, initieel slijm en dan etterachtig. Geleidelijk aan wordt een klinisch beeld gevormd van chronische obstructieve bronchitis en bronchiëctasie, emfyseem van de longen en ademhalingsfalen. Kinderen zijn buitengewoon gevoelig voor acute respiratoire-virale en bacteriële infecties, wat bijdraagt aan exacerbaties en progressie van bronchopulmonale pathologie. Mogelijk de ontwikkeling van infectieus afhankelijk bronchiaal astma.
Bij kinderen in de leerplichtige leeftijd kan mucoviscidose zich manifesteren als "darmkoliek". Patiënten klagen over ernstige paroxysmale pijnen in de buik, opgeblazen gevoel, herhaaldelijk braken. Bij het palperen van de buik, worden dichte formaties geïdentificeerd, gelegen in de projectie van de dikke darm - uitwerpselen, vermengd met dik, dicht slijm. Kinderen zijn erg vatbaar voor de ontwikkeling van hypochloremische alkalose als gevolg van overmatig zoutverwijdering met zweet bij warm weer, terwijl "zoutvorst" op de huid van het kind verschijnt.
De nederlaag van het bronchopulmonale systeem bij volwassenen
Het verslaan van bronchopulmonale systeem bij patiënten met cystische fibrose (pulmonaire vorm van de ziekte) wordt gekenmerkt door de ontwikkeling van chronische etterige obstructieve bronchitis, bronchiëctasie, chronische longontsteking, emfyseem, respiratoire insufficiëntie, cor pulmonale. Sommige patiënten ontwikkelden pneumothorax en andere complicaties van cystic fibrosis: atelectase, longabces, bloedspuwing, pulmonale bloeding, infectie-afhankelijke astma.
Patiënten klagen over een pijnlijke paroxysmale hoest met een zeer viskeus, moeilijk te scheiden mucopurulent sputum, soms met een bijmenging van bloed. Bovendien is dyspnoe in het begin zeer kenmerkend onder fysieke stress en vervolgens in rust. Dyspnoe is te wijten aan bronchiale obstructie. Veel patiënten klagen over chronische rhinitis veroorzaakt door polyposis en sinuititis. Er is ook een significante zwakte, een progressieve achteruitgang in de prestaties, frequente acute respiratoire-virale ziekten. Bij onderzoek wordt aandacht besteed aan de bleekheid van de huid, wallen in het gezicht, cyanose van de zichtbare slijmvliezen, uitgesproken dyspneu. Met de ontwikkeling van een gedecompenseerd pulmonaal hart verschijnt oedeem op de benen. Er kan verdikking zijn van de terminale vingerkootjes van de vingers van de handen in de vorm van trommelstokken en nagels - in de vorm van zandlopers. De thorax krijgt een tonvormige vorm (door de ontwikkeling van emfyseem).
Wanneer percussie van de longen zijn bepaalde tekenen van emfyseem - boxgeluid, een scherpe beperking van de beweeglijkheid van de pulmonale rand, het verlagen van de onderste rand van de longen. Met auscultatie van de longen, harde ademhaling met langdurige uitademing wordt onthuld, verspreide droge piepgeluiden, vochtig medium en kleine borrelende rales. Met een uitgesproken emfyseem van de longen wordt de ademhaling sterk verzwakt.
Extrathoracale manifestaties van cystische fibrose
Extrapulmonaire manifestaties van cystische fibrose kunnen behoorlijk uitgesproken zijn en komen vaak voor.
Versla de pancreas
Insufficiëntie van de exocriene functie van de pancreas in verschillende mate wordt waargenomen bij 85% van de patiënten met cystic fibrosis. Met een lichte beschadiging pancreas maldigestie en malabsorptiesyndromen afwezig, er enige laboratorium manifestaties exocriene insufficiëntie (lage trypsine en lipase in het bloed en duodenale inhoud, vaak uitgedrukt steatorrhea). Het is bekend dat om het syndroom van maldigestie te voorkomen slechts 1 tot 2% van het totale lipase wordt uitgescheiden. Klinisch worden alleen significante schendingen van de externe secretoire functie zichtbaar.
Onder normale omstandigheden wordt de afscheiding van een vloeibare consistentie rijk aan enzymen geproduceerd in de acini van de pancreas. Met de voortgang van de afscheiding via het uitscheidingskanaal, is het verrijkt met water en anionen, en wordt het nog meer vloeibaar. Bij cystic fibrosis gevolg van verstoring van de structuur en functie van transmembraan regulator (chloridekanalen) in de pancreas bevat is niet ontvangen voldoende vloeistof viskeus en de snelheid van de voortgang ductless vertraagt drastisch. Geheime eiwitten worden afgezet op de wanden van kleine uitscheidingskanalen, waardoor hun obstructie zich ontwikkelt. Naarmate de ziekte vordert, ontwikkelen zich uiteindelijk afbraak en atrofie van de acini - chronische pancreatitis met exocriene pancreasinsufficiëntie wordt gevormd. Dit wordt klinisch weerspiegeld in de ontwikkeling van syndromen van maldigestie en malabsorptie. Pancreatische insufficiëntie is de hoofdoorzaak van malabsorptie van vet bij cystische fibrose, maar in de regel wordt dit waargenomen bij een significante lipasedeficiëntie. Forsher en Durie (1991) geven aan dat bij afwezigheid van pancreatische lipase en geabsorbeerd vet wordt gesplitst met 50-60% door de aanwezigheid van maag en speekselklier (sublinguale) lipaseactiviteit dat dicht bij de ondergrens van normaal. Samen met de schending van de splitsing en opname van vetten, is er een overtreding van de splitsing en reabsorptie van eiwitten. Bij uitwerpselen gaat ongeveer 50% van het met voedsel gevoede eiwit verloren. Absorptie van koolhydraten lijdt minder, ondanks het tekort aan α-amylase, maar het metabolisme van koolhydraten kan aanzienlijk worden verminderd.
De nederlaag van de alvleesklier komt tot uiting in de ontwikkeling van het syndroom van maldigestie en malabsorptie met aanzienlijk gewichtsverlies, overvloedige vette ontlasting.
De ontwikkeling van syndromen van maldigestie en malabsorptie wordt ook vergemakkelijkt door ernstige aantasting van de functie van de darmklieren, een overtreding van de afscheiding van darmsap en een vermindering van het gehalte aan darmzymen daarin.
Syndromen van maldigestie en malabsorptie worden ook de darmvorm van cystische fibrose genoemd.
Overtreding van de incrementele functie van de pancreas (diabetes mellitus) wordt waargenomen bij patiënten met cystische fibrose in de late stadia van de ziekte (bij 2% van de kinderen en bij 15% van de volwassenen)
Laesie van de lever en galwegen
Bij 13% van de patiënten met gemengde en intestinale cystische fibrose ontwikkelt zich cirrose. Het is het meest typerend voor mutaties van W128X, delta-P508 en X1303K. Bij 5-10% van de patiënten wordt biliaire cirrose met portale hypertensie gevonden. Volgens Welch, Smith (1995) worden bij 86% van de patiënten met cystische fibrose klinische, morfologische, laboratorium- en instrumentele tekenen van leverbeschadiging gevonden.
Veel patiënten met cystische fibrose ontwikkelen ook chronische cholecystitis, vaak een rekenfout.
Overtreding van de functie van de seksuele klieren
Bij patiënten met cystische fibrose kan azoospermie optreden, wat de oorzaak is van onvruchtbaarheid. Verminderde vruchtbaarheid is ook kenmerkend voor vrouwen.
Stages
Er zijn drie graden van ernst van de longvorm van cystische fibrose.
De milde vorm van cystische fibrose wordt gekenmerkt door zeldzame exacerbaties (niet vaker dan eenmaal per jaar), tijdens perioden van remissie zijn klinische manifestaties praktisch afwezig, de zieken kunnen werken.
Het beloop van matige ernst - exacerbatie wordt 2-3 keer per jaar waargenomen en duurt ongeveer 2 maanden en langer. In de acute fase wordt intense hoest met moeilijk te scheiden sputum waargenomen, kortademigheid, zelfs bij onbeduidende lichamelijke inspanning, subfebrile lichaamstemperatuur, algemene zwakte, zweten. Tegelijkertijd is er een schending van de exocriene functie van de alvleesklier. In de fase van kwijtschelding is de arbeidscapaciteit niet volledig hersteld, dyspnoe wordt behouden tijdens lichamelijke inspanning.
Een ernstig verloop wordt gekenmerkt door zeer frequente exacerbaties van de ziekte. Er zijn praktisch geen remissies. In het klinische beeld is ernstig ademhalingsfalen, symptomatologie van het chronische longhart, vaak gedecompenseerd, gekenmerkt door bloedspuwing, in de voorhoede. Er is een aanzienlijke afname van het lichaamsgewicht, de patiënten zijn volledig uitgeschakeld. In de regel gaat ernstige bronchopulmonale pathologie gepaard met een uitgesproken stoornis van de pancreasfunctie.
Vormen
- Broncho-pulmonaire laesies
- Herhaalde en terugkerende pneumonie met langdurige loop.
- Abnormale pneumonie, vooral bij zuigelingen.
- Chronische pneumonie, vooral bilateraal.
- Bronchiaal astma, ongevoelig voor conventionele therapie.
- Terugkerende bronchitis, bronchiolitis, vooral met het zaaien van Pseudomonas aeruginosa.
- Veranderingen in het maagdarmkanaal
- Meconial ileus en zijn equivalenten.
- Syndroom van verminderde intestinale absorptie van onbekende oorsprong.
- Geelzuchtige obstructieve type bij pasgeborenen met langdurige loop.
- Cirrose van de lever.
- Diabetes mellitus.
- Gastro-oesofageale reflux.
- Cholelithiasis.
- Verzakking van het rectum.
- Veranderingen in andere organen en systemen
- Aandoeningen van groei en ontwikkeling.
- Vertraagde seksuele ontwikkeling.
- Mannelijke onvruchtbaarheid.
- Poliepen van de neus.
- Sibs uit families met patiënten met cystic fibrosis.
[24]
Complicaties en gevolgen
Complicaties uit het maagdarmkanaal omvatten:
- Diabetes mellitus ontwikkelt zich bij 8-12% van de patiënten ouder dan 25 jaar.
- Fibrotische colonopathie.
- Meconiumileus in de neonatale periode (12% van de kinderen met CF, het distale intestinale obstructie syndroom, rectale prolaps, ulcus pepticum en gastro-oesofageale refluxziekte.
Complicaties van de lever:
- Vettige degeneratie van de lever (bij 30-60% van de patiënten),
- Focal biliaire cirrose, multinodulaire biliaire cirrose en bijbehorende portale hypertensie.
Portale hypertensie leidt soms tot de dood als gevolg van spataderen van de slokdarm.
De prevalentie van cholecystitis en cholelithiase is hoger bij patiënten met cystische fibrose dan bij andere personen.
Vertraagde puberteit en verminderde vruchtbaarheid en andere complicaties. De meeste mannen hebben azoospermie en onderontwikkeling van de zaadleider.
Diagnostics cystische fibrose
Een gebruikelijke bloedtest wordt gekenmerkt door bloedarmoede van verschillende ernst, meestal normo- of hypochroom. Bloedarmoede heeft een multifactoriële genese (afname van de absorptie van ijzer en vitamine B12 in de darm als gevolg van de ontwikkeling van het malabsorptiesyndroom). Mogelijke leukopenie, met de ontwikkeling van purulente bronchitis en pneumonie - leukocytose, een toename van de ESR.
De algemene analyse van urine - zonder significante veranderingen, in zeldzame gevallen is er sprake van onbeduidende proteïnurie.
Coprologisch onderzoek - er is steatorrhoea, creatorrhea. Becker (1987) beveelt meting in de feces van chymotrypsine en vetzuren aan. Voordat de chymotrypsine in de ontlasting wordt bepaald, moet de inname van spijsverteringsenzymen niet minder dan 3 dagen vóór de test worden geannuleerd. Bij cystic fibrosis is de hoeveelheid chymotrypsine in de feces verminderd en is het aantal vetzuren verhoogd (de normale afgifte van vetzuren is minder dan 20 mmol / dag). Er moet rekening worden gehouden met het feit dat een verhoogde excretie met feces van vetzuren ook wordt waargenomen wanneer:
- tekort aan geconjugeerde vetzuren in de dunne darm met leverinsufficiëntie, obstructie van de galwegen, significante bacteriële kolonisatie van de dunne darm (intensieve hydrolyse van galzuren optreedt);
- ileitis;
- coeliakie (met de ontwikkeling van malabsorptiesyndroom);
- enteritis;
- intestinale lymfomen;
- De ziekte van Whipple;
- voedselallergieën;
- versnelde doorvoer van voedselmassa's voor diarree met verschillende genese, carcinoïdesyndroom, thyreotoxicose.
Biochemische analyse van bloed - vermindering van de totale hoeveelheid eiwit en albumine te verhogen in alfa 2 en gamma-globuline, bilirubine en transaminase (leverziekte), verminderde activiteit van amylase, lipase, trypsine en ijzergehalte van calcium (in ontwikkeling syndroom maldigestie, malabsorptie).
Sputum-analyse - de aanwezigheid van een groot aantal neutrofiele leukocyten en micro-organismen (met sputumuitstrijkje).
Onderzoek naar de absorptiefunctie van de dunne darm en de exocriene functie van de pancreas - significante schendingen worden gedetecteerd.
Röntgenonderzoek van de longen - onthult veranderingen, waarvan de ernst afhangt van de ernst en fase van de ziekte. De meest karakteristieke veranderingen zijn:
- toename van de intensiteit van het pulmonaire patroon als gevolg van peribronchiale interstitiële veranderingen;
- uitbreiding van de wortels van de longen;
- patroon van lobulaire, subsegmentale of zelfs segmentale atelectase van de longen;
- verhoogde transparantie van pulmonale velden, voornamelijk in de bovenste delen, laagblijvende en onvoldoende diafragma-mobiliteit, verwijding van de vaginale ruimte (manifestatie van emfyseem van de longen);
- segmentale of polysegmentaire infiltratie van longweefsel (met de ontwikkeling van pneumonie).
Bronchografie - detecteren veranderingen veroorzaakt tie obstructieve bronchiale viskeuze sputum (bronchiale fragmentatie vulling daarentegen ongelijke contouren bronchiën breuk fenomeen, een significante daling van het aantal zijtakken) en bronhoekgazy (cilindrische, gemengd) voornamelijk gelokaliseerd in de lagere longen.
Bronchoscopie - detecteert een diffuse purulente bronchitis met een overvloedige hoeveelheid dik, stroperig sputum en fibrineuze films.
Spirography - zelfs in de vroege stadia van de ziekte onthult de respiratoire soort falen (vermindering van FVC, FEV1 index Tiffno), beperking (verminderde VC) of vaker obstructieve-beperkende (verminderde vitale capaciteit, FVC, FEV1, Tiffno index).
Een zweettest (een studie van zweetelektrolyten) door Gibson en Cook - stimulatie van zweten met behulp van elektroforese met pilocarpine gevolgd door bepaling in het zweetchloride. Doerehuk (1987) beschrijft het monster als volgt. Elektroforese pilocarpine wordt geproduceerd in het gebied van de onderarm, de elektrische stroom is 3 mA. Nadat de huid met gedestilleerd water is gereinigd, wordt zweet verzameld met filtreerpapier op het gestimuleerde gebied, bedekt met gaas om verdamping ervan te voorkomen. Na 30-60 minuten wordt het filterpapier verwijderd en geëlueerd in gedestilleerd water. Meet de hoeveelheid verzameld zweet. Voor het verkrijgen van betrouwbare resultaten is het noodzakelijk om ten minste 50 mg (bij voorkeur 100 mg) zweet te verzamelen.
Bij een chlorideconcentratie van meer dan 60 mmol / l wordt de diagnose van cystische fibrose waarschijnlijk geacht; bij een chlorideconcentratie van meer dan 100 mmol / l - betrouwbaar; terwijl het verschil in de concentratie van chloor en natrium niet hoger mag zijn dan 8-10 mmol / l. Hadson (1983) beveelt een monster met prednisolon aan op de grenswaarde van natrium- en chloridegehalte (2 dagen oraal 5 mg innemen, gevolgd door elektrolyten in het zweet). Bij personen die geen cystische fibrose hebben, neemt het natriumgehalte in het zweet af tot de waarde van de onderste limiet van de norm, met cystische fibrose - het verandert niet. Een zweterig monster wordt aanbevolen voor elk kind met een chronische hoest.
De analyse van bloedvlekken of DNA-monsters op belangrijke mutaties van het cystic fibrosis-gen is de meest gevoelige en specifieke diagnostische test. Deze methode is echter geschikt voor landen waar de mutatiefrequentie van delta-P508 hoger is dan 80%. Bovendien is de techniek erg duur en technisch ingewikkeld.
Prenatale diagnose van cystic fibrosis - wordt gedaan door de isoenzymen van alkalische fosfatase in het vruchtwater te bepalen. Deze methode is mogelijk met 18-20 weken zwangerschap.
De belangrijkste criteria voor de diagnose van cystische fibrose zijn de volgende:
- indicaties in de geschiedenis van de kindervertraging bij lichamelijke ontwikkeling, terugkerende chronische aandoeningen van de luchtwegen, dyspeptische stoornissen en diarree, de aanwezigheid van cystische fibrose bij naaste familieleden;
- chronische obstructieve bronchitis, vaak recidiverend, met de ontwikkeling van bronchiëctasie en emfyseem, vaak terugkerende pneumonie;
- chronische recidiverende pancreatitis met duidelijke afname van de exocriene functie, malabsorptiesyndroom;
- verhoogd chloorgehalte in het zweet van de patiënt;
- onvruchtbaarheid met bewaarde seksuele functie.
Succesvolle diagnose en differentiële diagnose van cystic fibrosis wordt vergemakkelijkt door de identificatie van risicogroepen.
Het programma van onderzoek voor cystic fibrosis
- Algemene analyse van bloed, urine, sputum.
- Bacteriologische analyse van sputum.
- Coprologische analyse.
- Biochemische analyse van bloed: bepaling van totaal eiwit en eiwitfracties, glucose, bilirubine, transaminasen, alkalische fosfatase, gamma-glutamyltranspeptidase, kalium, calcium, ijzer, lipase, amylase, trypsine.
- Onderzoek van de exocriene functie van de pancreas en de intestinale absorptiefunctie.
- Röntgen- en longradiografie, long-CT.
- ECG.
- Echocardiografie.
- Bronchoscopie en bronchografie.
- Spirography.
- Een zweettest.
- Raadpleging van een geneticus.
- Analyse van bloedvlekken of DNA-monsters voor belangrijke mutaties van het cystic fibrosis-gen.
Wat moeten we onderzoeken?
Welke tests zijn nodig?
Met wie kun je contact opnemen?
Behandeling cystische fibrose
Het type en de ernst van de symptomen van cystische fibrose kunnen erg verschillen, dus er is geen standaard behandelplan, het is in elk geval individueel.
Therapie bestaat uit de volgende behandelingsmaatregelen:
- Ademhalingsoefeningen en houdingsdrainage, helpen om zich te ontdoen van het dikke slijm dat zich ophoopt in de longen. Sommige methoden voor luchtwegzuivering vereisen hulp van familieleden, vrienden of een longarts. Veel mensen gebruiken een opblaasbaar borstvest dat trilt met een hoge frequentie.

- Inhalatie medicijnen die bronchodilatator, drainage (mucolytica) en antibacteriële effecten (bijv. Fluorochinolonen) uitoefenen.
- Preparaten die pancreasenzymen bevatten om de spijsvertering te verbeteren. Deze medicijnen worden ingenomen met voedsel.
- Multivitaminen (inclusief in vet oplosbare vitamines).
In 2015 keurde de FDA een tweede medicijn goed voor de behandeling van cystische fibrose, dat een defect eiwit als CFTR aantast. Het eerste medicijn, de zogenaamde CFTR-modulator, werd in 2012 goedgekeurd. Verwacht wordt dat CFTR-modulatoren het leven van sommige mensen met cystische fibrose gedurende tientallen jaren kunnen verlengen.
Chirurgische behandeling kan nodig zijn om de volgende respiratoire complicaties te behandelen:
- Pneumothorax, massieve terugkerende of aanhoudende bloedspuwing, neuspoliepen, aanhoudende en chronische sinusitis.
- Mekonische obstructie, intussusceptie, prolaps van rectum.
Longtransplantatie wordt uitgevoerd in het terminale stadium van de ziekte.
Prognose
De gemiddelde leeftijd van overleven van patiënten met cystic fibrosis varieert van 35 tot 40 jaar. De gemiddelde leeftijd van overleven bij mannen is hoger dan die van vrouwen.
Dankzij moderne behandelingsstrategieën bereikt 80% van de patiënten de volwassenheid. Niettemin beperkt cystische fibrose de functionele mogelijkheden van de patiënt aanzienlijk. Het medicijn voor deze ziekte is nog niet ontwikkeld.