Medisch expert van het artikel

Nieuwe publicaties
Prionen - veroorzakers van prionziekten
Laatst beoordeeld: 23.04.2024

Alle iLive-inhoud wordt medisch beoordeeld of gecontroleerd op feiten om zo veel mogelijk feitelijke nauwkeurigheid te waarborgen.
We hebben strikte richtlijnen voor sourcing en koppelen alleen aan gerenommeerde mediasites, academische onderzoeksinstellingen en, waar mogelijk, medisch getoetste onderzoeken. Merk op dat de nummers tussen haakjes ([1], [2], etc.) klikbare links naar deze studies zijn.
Als u van mening bent dat onze inhoud onjuist, verouderd of anderszins twijfelachtig is, selecteert u deze en drukt u op Ctrl + Enter.
Langzame virale infecties worden gekenmerkt door speciale criteria:
- ongewoon lange incubatietijd (maanden, jaren);
- een soort nederlaag van organen en weefsels, voornamelijk het centrale zenuwstelsel;
- langzame gestage progressie van de ziekte;
- een onvermijdelijke dodelijke afloop.

Sommige pathogenen van acute virale infecties kunnen ook langzame virale infecties veroorzaken. Het mazelenvirus veroorzaakt bijvoorbeeld soms PSES en het rodehondvirus - progressieve congenitale rubella en rodehond panencefalitis.
Een typische langzame virale infectie van dieren wordt veroorzaakt door virusvisna / medi, dat tot retrovirussen behoort . Het is de veroorzaker van langzame virale infectie en progressieve longontsteking van schapen. De witte massa van de hersenen wordt vernietigd , verlamming ontwikkelt zich (visna-verwelking); er is een chronische ontsteking van de longen en milt.
Soortgelijke ziekten als gevolg van langzame virusinfecties veroorzaken prionen - pathogenen van prionbesmettingen. Prionziekten zijn een groep van progressieve stoornissen van het menselijke en dierlijke CZS. Mensen hebben de functie van het centrale zenuwstelsel verstoord, er zijn veranderingen in persoonlijkheid, bewegingsstoornissen. Symptomen van de ziekte duren meestal van enkele maanden tot meerdere jaren en eindigen fataal. Eerder werden prioninfecties overwogen samen met de zogenaamde pathogenen van langzame virale infecties.
Sommige middelen die prionziekten veroorzaken, worden eerst in lymfoïde weefsels geaccumuleerd. Prionen die in de hersenen, accumuleert in grote hoeveelheden, waardoor amyloïdose (extracellulair disproteinoz gekenmerkt door amyloïde afzetting bij de ontwikkeling van atrofie en sclerose van weefsel) en astrocytose (astrocytische glia proliferatie, giperprodukdiya glialnyh vezels). Fibrils of amyloïde eiwitaggregaten en hersenen spongiforme veranderingen (overdraagbare spongiforme encefalopathie). Dientengevolge, gedragsveranderingen, coördinatie van bewegingen wordt verstoord, uitputting met dodelijke afloop ontwikkelt zich. Immuniteit wordt niet gevormd. Prion ziekten gerelateerd aan conformationele ziekten die ontstaan als gevolg van misfolding (schending van juiste conformatie) van cellulair eiwit nodig is voor de normale werking van het lichaam. De manieren om prionen over te brengen zijn divers:
- voedingsroute - geïnfecteerde producten van dierlijke oorsprong, voedingssupplementen van onbewerkte runderorganen, enz.:
- overdracht met bloedtransfusie, toediening van dierlijke producten, transplantatie van organen en weefsels, gebruik van geïnfecteerde chirurgische en tandheelkundige instrumenten;
- overdracht via immunobiologische geneesmiddelen (waarvan bekend is dat deze PrpP '' '1500 schapenhersenen formulvaccine van zieke schapen infecteren).
Pathologische prionen, die in de ingewanden zijn geraakt, worden getransporteerd naar het bloed en de lymfe. Na perifere replicatie in de milt, appendix, amandelen en andere lymfoïde weefsels, worden ze via perifere zenuwen (neuro-invasie) naar de hersenen overgebracht. Waarschijnlijk directe penetratie van prionen in de hersenen via de bloed-hersenbarrière. Eerder werd aangenomen dat het centrale zenuwstelsel het enige weefsel is waarin pathologische prionen accumuleren, maar er zijn onderzoeken naar voren gekomen die deze hypothese hebben veranderd. Het bleek dat de ophoping van prionen in de milt gepaard gaat met de toename en het functioneren van folliculaire dendritische cellen.
 [
[Eigenschappen van prionen
De cellulaire normale isovorm van prioneiwit met een molecuulgewicht van 33-35 kD wordt bepaald door het prioneiwit-gen (prion-gen PrNP bevindt zich op het 20ste chromosoom van een mens). Een normaal gen verschijnt op het oppervlak van de cel (verankerd in het membraan door het glycoproteïne-molecuul), is gevoelig voor het protease. Het reguleert de overdracht van zenuwimpulsen, dagcycli, oxidatieprocessen, participeert in het metabolisme van koper in het centrale zenuwstelsel en in de regulatie van de verdeling van beenmergstamcellen. Bovendien wordt het prion-gen gevonden in de milt-, lymfeknopen-, huid-, GIT- en folliculaire dendritische cellen.
Proliferatie van pathologische prionen
De transformatie van prionen in gewijzigde vormen vindt plaats wanneer het kinetisch geregelde evenwicht tussen hen wordt verstoord. Het proces wordt versterkt wanneer de hoeveelheid pathologisch (PrF) of exogeen prion toeneemt. PrP is een normaal eiwit verankerd in het celmembraan. PrP 'is een globulair hydrofoob eiwit, dat aggregaten met zichzelf en met PrF "op het oppervlak van de cel vormt: als een resultaat wordt PrP' omgezet in PrF" en de cyclus wordt voortgezet. De pathologische vorm van PrF "" stapelt zich op in neuronen, waardoor de cel een sponsachtig uiterlijk krijgt.
Kuru
Prionziekte, voorheen veel voorkomend onder de Papua's (vertaald als trillen of beven) in het oostelijke deel van het eiland Nieuw-Guinea. Besmettelijke eigenschappen van de ziekte bewezen K. Gaidushek. Het veroorzakende agens wordt door voedsel overgedragen als gevolg van rituele kannibalisme - het eten van onvoldoende thermisch verwerkte, geïnfecteerde prionen van de naaste familieleden van de hersenen. Als gevolg van de nederlaag van het centrale zenuwstelsel, zijn bewegingen, gang, rillingen, euforie ("lachende dood") verstoord. De incubatieperiode duurt 5-30 jaar. Een jaar later sterft de patiënt.
Ziekte van Creutzfeldt-Jakob
Prionziekte, die in de klassieke variant van de ziekte van Creutzfeldt-Jakob en stroomt in de vorm van dementie, visuele en cerebellaire stoornissen en bewegingsstoornissen fatale ziekte bij 4-5 maanden (3-14 maanden wanneer nieuwe variant van de ziekte van Creutzfeld-Jakob. De incubatietijd kan oplopen 20. Er zijn verschillende manieren van infectie en de oorzaken van de ziekte:
- bij gebruik van onvoldoende thermisch verwerkte producten van dierlijke oorsprong, bijvoorbeeld vlees, hersenkoeien, patiënten met boviene spongiforme encefalopathie;
- voor de transplantatie van weefsels, bijvoorbeeld het hoornvlies van het oog, bloedtransfusie, het gebruik van hormonen en andere biologisch actieve stoffen van dierlijke oorsprong, het gebruik van catgut, gecontamineerde of onvoldoende gesteriliseerde chirurgische instrumenten, prozectiemanipulaties;
- voor hyperproductie van PrP en andere staten die het proces van transformatie van PrP 'in PrF stimuleren. "
De ziekte kan zich ook ontwikkelen als een resultaat van een mutatie of insertie in het prion-genregio. Het familiekarakter van de ziekte is wijdverspreid als gevolg van een genetische aanleg voor de ziekte van Creutzfeldt-Jakob. Met een nieuwe variant van de ziekte van Creutzfeldt-Jakob ontwikkelen zich aandoeningen op een jongere leeftijd (gemiddelde leeftijd 28 jaar), in tegenstelling tot de klassieke variant (gemiddelde leeftijd 65 jaar). Met de nieuwe variant van de ziekte van Creutzfeldt-Jakob accumuleert abnormaal prionisch eiwit niet alleen in het centrale zenuwstelsel, maar ook in lymforeticulaire weefsels, inclusief in de amandelen.
Het syndroom van Gerstmann-Streussler-Sheinker
Erfelijke prionziekte, die optreedt met dementie, hypotensie, slikken (dysfagie), dysartrie. Heeft vaak een familiekarakter. De incubatietijd is van 5 tot 30 jaar. De ziekte komt voor in 50-60 jaar, de duur varieert van 5 tot 13 jaar.
Erfelijke sterfelijke slapeloosheid
Auto-immuunziekte met voortschrijdende slapeloosheid, sympathische hyperreactiviteit (hypertensie, hyperthermie, hyperhidrose, tachycardie), tremor, ataxie, veellettergrepels, hallucinaties. De slaap is sterk gestoord. Dood treedt op met de progressie van cardiovasculair falen.
Repareer het
Scrapie (uit het Engels schrapen -. Scrape) - prionziekte van schapen en geiten (schurft), stroomt met de ziekte van het centrale zenuwstelsel, progressieve aandoening van de beweging, sterke pruritus (jeuk), en eindigt in de dood van het dier.
Boviene spongiforme encefalopathie
Ziekte van vee, gekenmerkt door de nederlaag van het centrale zenuwstelsel, een schending van coördinatie van bewegingen en de onvermijdelijke dood van het dier. Voor het eerst brak de epidemie van de ziekte uit in het VK. Het werd geassocieerd met het voederen van dieren met vlees- en beendermeel dat pathologische prionen bevatte. De incubatietijd varieert van 1,5 tot 15 jaar. De meest geïnfecteerde zijn het hoofd, het ruggenmerg en de oogbollen van dieren.
Laboratoriumdiagnostiek van prionziekten
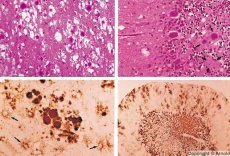
Bij diagnose spongiforme veranderingen in de hersenen, astrocytose (gliosis), afwezigheid van infiltraten van ontsteking. De hersenen zijn gekleurd met amyloïde. In het cerebrospinale vocht worden eiwitmarkers van prion-hersenaandoeningen gedetecteerd (met behulp van ELISA). Genetische analyse van prion-gen (PCR) wordt uitgevoerd.
Profylaxe van prionenziekten
Voor decontaminatie van gereedschappen en milieu objecten aanbevolen autoclaaf (bij 134 ° C 18 min bij 121 ° C gedurende 1 uur)., Brandende, extra bleken verwerking en odnonormalnym oplossing van NaCl in 1 uur voor niet-specifieke preventie beperkingen oplegt aan het gebruik van geneesmiddelen van dierlijke oorsprong en de productie van hypofysehormonen van dierlijke oorsprong is verboden. Beperk de transplantatie van de dura mater. Bij het werken met de dialogische vloeistoffen van patiënten met rubberen handschoenen.